微生物ms17-010win10
ms17-010win10 时间:2021-05-20 阅读:()
微生物菌群高通量测序www.
genewiz.
com.
cn微生物菌群高通量测序1.
研究背景苏州金唯智生物科技有限公司Project.
Asia@genewiz.
com.
cn400-8100-669转3简介发布时间:2016/4/1关键词:高通量测序微生物菌群多样性16S18SITS高通量测序技术的出现已经彻底改变了微生物的多样性研究.
采用高通量测序手段,对栖息在多种多样的生态系统(例如肠道和极端环境)中的微生物群落进行检测,能够突破传统微生物分离培养和显微观察等手段对微生物检测的限制,同时研究不可培养或无法观察的未知微生物,统计微生物群体的多样性与丰度,分析微生物群体基因及功能,探求微生物与环境/宿主之间的关系,发掘和研究新的功能基因.
微生物组研究有望改善人类健康、农业、生物能源和环境,也是目前发展最迅猛的研究领域之一.
金唯智公司提供16SMetaVx测序、18S测序和ITS测序服务.
本文重点介绍高通量测序在微生物菌群检测中的应用,Meta16S/18S/ITS测序实验流程、分析重点、适用范围和注意事项等.
摘要2.
材料和方法Meta16S/18S/ITS测序可以针对土壤、粪便、空气、水体、动植物体表和体内等任何来源提取的全基因组DNA进行检测.
16SMetaVx测序DNA总量大于70ng,浓度大于2ng/ul即可满足扩增的需要.
18S测序和ITS测序则建议DNA总量大于100ng,浓度大于5ng/ul.
宏基因组样品受样品来源和杂质含量等影响较大,样品中2.
1实验材料自然状态下,微生物几乎无处不在.
土壤、海洋、肠道等生态系统中的微生物数量繁多、种类多样,是环境能量、物质代谢的重要中间环节和组成部分.
微生物虽然结构简单,体积微小,却有着重要意义,因其在工业、农业、医疗卫生、环境保护等各方面的重要地位,被越来越多的研究者关注[1,2].
以往研究环境微生物的手段主要基于微生物分离培养和显微观察等.
传统的培养方法只限于对环境样品中极少部分(0.
1%-1%)可培养的微生物类群的研究[3];显微观察受分辨率的限制,无法观测到体积较小的微型浮游动植物和细菌等,含量较低的生物也很难观测;而变性梯度凝胶电泳(DGGE)、克隆文库等常规的分子生物学方法也因操作复杂、成本高、痕量菌发现困难等因素无法达到深入分析环境微生物多样性的目的[4].
高通量测序技术的出现,使每个样品的测序量达到普通测序的几百倍,而单条序列的测序费用远低于普通测序.
它极大的促进了对环境中不可培养微生物以及痕量菌的研究,为环境微生物多样性的研究开启了新的研究热潮[5].
高通量测序技术的出现摆脱了传统研究方法的限制,它可以对环境微生物的DNA直接进行深度测序,进而分析环境微生物的群落结构、系统进化、基因功能差异及代谢网络等,这使得微生物菌群检测日益成为新的研究热点[6].
高通量测序在宏基因组学研究中的应用主要有两方面:一是对所有微生物DNA进行宏基因组测序,关注样品中菌群组成、系统进化和功能基因的研究和发掘;二是基于16SrDNA,18srDNA,ITS区域进行扩增测序,分析微生物的群体构成和多样性,探求微生物与环境,微生物与宿主之间的关系等.
后者针对特异区域进行PCR扩增并测序,由于操作简便,仅需很少的数据量即可提供环境样本物种分类,物种丰度,种群结构,系统进化,群落比较等诸多信息,在微生物分类鉴定,微生态研究方等方面起到重要作用[7].
16SMetaVx是金唯智公司独立研发的微生物菌群高通量测序平台,多套引物同时扩增细菌和古细菌的V3、V4和V5三个可变区,真实反映菌群组成;MiSeq(PE300)测序,每个样品不低于5万条reads,能在短时间内获得全面和准确的微生物群落信息,增强对痕量微生物的检出[8-10].
本文根据金唯智的16SMetaVx测序服务,详细阐述高通量测序在微生物种群多样性检测中的实验流程、检测效果和注意事项等.
细菌基因组的含量、基因组中腐植酸的残留都会对后续的扩增产生使用Qubit2.
0(FluorometerInvitrogen,Carlsbad,CA)和0.
8%琼脂糖凝胶电泳对提取得到的DNA进行质检.
使用MetaVx文库构建试剂盒(GENEWIZ,Inc.
,SouthPlainfield,NJ,USA)构建测序文库,具体步骤是取5-50ngDNA,采用两步PCR扩增法扩增细菌和古菌16SrRNA的V3,V4和V5区.
设计带有"P5/P7-barcode-测序引物-特异引物"的融合引物,通过严格的PCR扩增体系和条件,构建测序文库.
使用Agilent2100Bioanalyzer(AgilentTechnologies,PaloAlto,CA,USA)对文库进行质检,带有不同barcode的文库通过定量和均一化,混合后使用IlluminaMiSeq(Illumina,SanDiego,CA,USA)测序仪进行300PE测序.
2.
2实验流程3.
结果和分析高通量测序可以得到大量的数据,为了从中找出需要的信息,发掘生物学意义,需要进行复杂和专业的生物信息学统计和分析.
这也是微生物菌群检测服务的重点.
图1微生物菌群检测项目流程3.
1分析流程基础分析高级分析物种百分比同级Top30的物种热图Top30的物种分布柱状图PCoA分析(Un)WeightedUnifrac分析UPGMATreeLEfSE分析RDA/CCA分析NMDS分析分类学系统组成树OTU分类学统计RankAbundance曲线OTU维恩图α多样性分析稀释性曲线OTUheatmap图序列长度及分布统计有效序列统计物种分类OTU分析物种丰度群落结构原始数据高质量序列样本聚类树与柱状图组合RDP0.
97数据预处理2OTU生成组间显著性差异metastats分析然后由MiSeq自带的MiSeqControlSoftware(MCS)读取序列信息.
某些样品来自和寄主共生的环境,如植物共生菌,昆虫的肠道微生物等,提取时可能包括了寄主本身的大量DNA,对DNA的总量要求会提高,建库流程也需要相应调整.
微生物菌群多样性检测受DNA提取和扩增影响很大,不同的扩增区段和扩增引物甚至PCR循环数的差异都会对结果有所影响.
因而建议同一项目不同样品都采用相同的条件和测序方法,这样相互之间才存在可比性.
影响.
因此需要尽量提高DNA纯度,260/280比值在1.
8-2.
2之间,琼脂糖电泳条带清晰无显著降解.
为了得到足够的基因组DNA,常规样品一般建议提供2g以上沉淀,或10ml以上水样,采用冰袋进行运输.
OTU是在群体遗传学研究中,为了便于分析,人为给某一个分类单元(属、种、分组等)设置的统一标志.
在生物信息分析中,测序得到的每一条序列来自于一个菌种,要了解一个样本测序结果中的菌种、属等数目信息,就需要对序列进行归类操作.
通过归类操作,将序列按照彼此的相似性归类为许多小组,一个小组就是一个OTU.
通常在97%的相似水平下对所有序列进行OTU划分并进行生物信息统计分析.
图3中不同颜色的圈表示不同的样本,图中的数字分别代表了每个样本(或分组)特有或共有的OTU数目.
图4中每条曲线对应一个样本,横坐标为OTU相对丰度含量按等级降序排列(OTURank),纵坐标为该OTU中序列数的相对百分含量(RelativeAbundance).
如横坐标100表示样本中按照相对丰度降序排列在第100的OTU,纵坐标为该等级OTU中序列数的相对百分含量(该OTU的序列数除以总序列数).
图5横坐标为样本名(SampleName),纵坐标为不同物种相对丰度(RelativeAbundance),图例为门水平的物种分类名称,Other表示除相对丰度前20外的所有其他门的相对丰度和.
柱状图中自下而上的颜色对应右边图例的颜色顺序.
图3样本OTUVenn图3.
2标准分析内容3.
2.
1测序数据初步统计和质量优化3.
2.
2OTU分析3.
2.
3物种丰度和物种均匀度分析图2有效序列长度分布图图4Rank-abundance曲线图5门水平的物种分类分析33.
2.
4物种分类分析β多样性分析需要3个或以上样品.
β多样性值为两个样本间的相异系数,反映不同样本间的多样性的差异,利用各样品序列间的进化和丰度信息计算样品间的距离,反映样品间是否有显著地微生物群落差异,可通过UniFrac分析实现.
样本聚类分析也需要3个及以上样品,利用各样品序列间的进化信息来比较环境样品在特定的进化谱系中是否有显著的微生物群落差异.
PCoA(PrincipalCo-ordinatesAnalysis)分析即主坐标分析,是一种研究数据相似性或差异性的可视化方法,它与PCA类似,通过一系列的特征值和特征向量进行排序后,选择主要排在前几位的特征值,找到距离矩阵中最主要的坐标,结果是数据矩阵的一个旋转,它没有改变样品点之间的相互位置关系,只是改变了坐标系统.
图6中PC1_vs_PC2为第一和第二主坐标得到的的PCoA图;PC1_vs_PC3为第一和第三主坐标得到的PCoA图;PC3_vs_PC2为第二和第三主坐标得到的PCoA图.
以PC1_vs_PC2为例,x轴与y轴分别代表第一和第二主坐标.
主坐标后的百分比代表此坐标对样本差异的贡献率,度量了此主坐标对原始信息量提取的多少.
样品点距离的远近代表了样品中微生物群落的相似性,距离越近,相似度越高;聚集在一起的样品由具有相似的微生物群落构成.
α多样性主要关注单样本的多样性分析,可以反映微生物群落中物种的数目,通过一系列统计学指数的分析来估计环境群落的物种丰度和多样性.
常用的度量指标有Chao1丰富度估计量(Chao1richnessestimator)、香农-威纳多样性指数(Shannon-wienerdiversityindex)、辛普森多样性指数(Simpsondiversityindex)等.
3.
2.
5α多样性分析图62DPCoAPlot图7UPGMATree3.
2.
6β多样性分析3.
2.
7PCoA分析3.
2.
8样本聚类分析4图7中每个分枝代表一个样本,内节点用不同颜色分出不同区间的bootstrap值.
红色表示bootstrap介于75-100%之间;黄色表示bootstrap值介于50-75%之间;绿色表示bootstrap值介于25-50%之间;蓝色表示bootstrap值小于25%.
非度量多维尺度法是一种将多维空间的研究对象简化到低维空间进行定位、分析和归类,同时又保留对象间原始关系的数据分析方法.
其特点是根据样品中包含的物种信息,以点的形式反映在多维空间上,而对不同样品间的差异程度,则是通过点到点间的距离体现的,最终获得样品的空间定位点图.
图8中不同颜色或形状的点代表了不同环境或条件下的样本.
RDA/CCA是基于对应分析发展而来的一种排序方法,将对应分析与多元回归分析相结合,每一步计算均与环境因子进行回归,又称多元直接梯度分析.
此分析是主要用来反映菌群与环境因子之间关系.
RDA是基于线性模型,CCA是基于单峰模型.
分析可以检测环境因子、样品、菌群三者之间的关系或者两两之间的关系.
图中数字表示样品名,不同颜色或形状表示不同环境或条件下的样本组;箭头表示环境因子;环境因子之间的夹角代表物种与环境因子间的正、负相关关系(锐角:正相关;钝角:负相关;直角:无相关性);由不同的样本向各环境因子做垂线,投影点越相近说明样本间该环境因子属性值越相似,即环境因子对样品的影响程度相当.
LefSe(LDAEffectSize)是一种用于识别高维生物标识和揭示基因组特征(包括基因,代谢和物种分类)的分析方法.
这些生物标识或基因组特征可以显示两个或两个以上生物条件(或者是类群)之间的差异.
3.
3高级数据分析图8NMDSPlot图10CCAPlot图9RDAPlot3.
3.
1NMDS分析3.
3.
2RDA/CCA分析3.
3.
3LEfSE分析5图11中A展示了两个组别中有显著作用的物种分类,并统计了LDA分析得到的分值;图B为物种聚类进化树,红色背景区域和绿色背景区域表示不同的分组,树枝中红色节点表示在红色组别中起到重要作用的微生物类群,绿色节点表示在绿色组别中起到重要作用的微生物类群,黄色节点表示的是在两组中均没有起到重要作用的微生物类群.
图中英文字母表示的物种名称在右侧图例中展示.
组间物种组成显著性差异分析根据不同组别的群落丰度数据,运用严格的统计学方法可以检测两组微生物群落中表现出丰度差异的分类,进行稀有频率数据的多重假设检验和假发现率(FDR)分析可以评估观察到的差异的显著性.
该分析可选择门、纲、目、科及属等不同分类学水平上进行.
图12中左图展示了样本UPGMA聚类树,右图展示了各样本的群落结构柱状图,不同颜色表示不同的物种分类,具体见左侧图例.
图13中A图为环形OTU进化树与OTU热图的组合分析展示,B为常规OTU进化树与OTU热图的组合分析展示.
图11LEfSEPlot图13MicrobialCommunityHeatmapwithOTUClusterTree3.
3.
6OTU/菌种进化树与热图组合分析3.
3.
4组间物种组成显著性差异分析AABB图12MicrobialCommunityBarplotwithSamplesClusterTree3.
3.
5样本聚类树与柱状图组合分析6rDNA(ribosomeDNA)指的是原/真核生物基因组中编码核糖体RNA(rRNA)分子对应的DNA序列,序列中既有保守区又有可变区,保守序列区域反映了生物物种间的亲缘关系,而高变序列区域则能体现物种间的差异.
16SrDNA是原核生物编码核糖体小亚基16SrRNA的基因,大小约1.
5Kb左右,是细菌的系统分类研究中最有用的和最常用的分子钟[7].
而18SrRNA(1855bp)的DNA序列是编码真核生物核糖体小亚基rRNA(18srDNA,1855bp)的DNA序列,由于在进化速率上比较保守,在系统发育研究中较适用于真核生物种级以上阶元的分类.
ITS(InternalTranscribedSpacer,内转录间隔区)是内转录间隔区是真菌核糖体RNA(rRNA)基因非转录组的一部分,ITS分为两个区域ITS1和ITS2.
5.
8s、18s、28srRNA基因上有极大的保守性,ITS不加入成熟核糖体,所以ITS片段在进化过程中承受的自然选择压力非常小,有更多的变异位点,通过检测ITS的序列变异和丰度,能反映环境样本中真菌的分类和丰度.
4.
问题和讨论图1416SMetaVxTMVs常规16S测序7GENEWIZ据此分别开发了针对微生物菌群多样性检测的Meta16S/18S/ITS测序平台,利用保守区设计引物将设计引物进行扩增测序,再根据可变区序列的差异来进行不同菌属的分类鉴定.
16SMetaVx测序主要鉴定细菌和古菌,适用于环境微生物、肠道菌群等样品的细菌鉴定;18S测序主要鉴定真菌及其他真核生物,适用于环境微生物、共生的真核生物菌群等;ITS测序主要鉴定真菌,如果样品以真菌和浮游微生物为主,或者仅关注样品中的真菌菌群,可以选择ITS测序.
相对于目前已有的宏基因组16S鉴定平台,金唯智开发的16SMetaVx测序平台兼具保守性和灵活性,能够覆盖绝大部分细菌菌群,并同时鉴定细菌和古菌.
比常规的16S检测更加灵敏,并且更能反映真实的菌群组成和结构(图14).
模拟测试样品,16SMetaVxTM平台检测出19个纲的微生物,而常规16S测序仅检测出11个纲.
*仅被16SMetaVxTM平台检出.
**占菌群80%左右,上图未包含.
图15增加V5区显著提高菌种检出率PhylumV3V4V5V3V4ClassOrderFamilyGenus0300600900物种数量物种分类层级8关于金唯智16S测序最适的数据量,取决于物种的丰度和对痕量微生物的检测要求.
一般情况下,物种丰度越高,或者对痕量微生物的检出水平要求越高,需要的数据量越大.
稀释曲线通过梯度抽取一定数量的有效序列,分析OTU检出情况.
随测序深度增加,OTU的数量随之增加.
当曲线趋于平缓时表示随着抽取的数据量的加大,检测到的OTU数目不再增加,此时的测序数据量较为合理.
为了平衡菌种检出率和测序成本,经过GENEWIZ长期验证,一般情况下每个文库测序5万条reads即可鉴定出微生物群落中绝大部分菌种(图16),此时性价相对较高.
图16横坐标为样本抽取的有效序列数(SequencePerSample),纵坐标为OTU数目(ObservedOTUs).
曲线解读:图中每条曲线代表一个样品,用不同颜色标记;随测序深度增加,OTU的数量随之增加.
综上,高通量测序已经给微生物菌群研究带来深远的影响.
金唯智公司开发的16S、18S和ITS测序,具有周期短、效率高、价格优惠、真实可信等优点,能够有效满足微生物菌群检测的需要.
图16稀释曲线图金唯智是专注于基因组研究和基因技术应用的生物技术公司,提供DNA测序、基因合成、高通量测序、引物合成、分子生物学服务、及GLP标准规范服务.
基于金唯智坚实的技术与优质的服务,包括近30位诺贝尔奖获得者在内的众多科研工作者已成为金唯智的忠实客户,全球诸多知名跨国公司以及著名高等学府也把金唯智选为其战略合作伙伴和首选供应商.
金唯智总公司(GENEWIZ)成立于1999年,总部位于美国新泽西州,另在美国马萨诸塞州、加利福尼亚州、华盛顿州、马里兰州、北卡罗来纳州、英国、日本以及中国的苏州、北京、天津设有公司.
其中苏州金唯智生物科技有限公司是金唯智在中国的总部.
经过金唯智人的共同努力,公司在其竞争的多项领域建立起行业领先的地位.
Kozich等评估了Miseq测序仪分析的不同16SrRNA可变区的准确性发现,测定V4区效果最佳[11].
进一步研究表明,在相同测序深度的情况下,采用16SMetaVxTM测序平台检测V3、V4和V5区,相比只检测v3和v4区,在不同物种分类层级上能多鉴定出10%~15%的菌种(图15).
9参考文献[1]ChristianN,WhitakerBK,ClayK(2015)Microbiomes:unifyinganimalandplantsystemsthroughthelensofcommunityecologytheory.
FrontMicrobiol6:869.
[2]DubilierN,McFall-NgaiM,ZhaoL(2015)Microbiology:Createaglobalmicrobiomeeffort.
Nature526:631-634.
[3]HaraszthyVI,ZambonJJ,SreenivasanPK,ZambonMM,GerberD,etal.
(2007)Identificationoforalbacterialspeciesassociatedwithhalitosis.
JAmDentAssoc138:1113-1120.
[4]CombesS,MichellandRJ,MonteilsV,CauquilL,SoulieV,etal.
(2011)Postnataldevelopmentoftherabbitcaecalmicrobiotacompositionandactivity.
FEMSMicrobiolEcol77:680-689.
[5]WangJ,WangF,ChuL,WangH,ZhongZ,etal.
(2014)HighgeneticdiversityandnoveltyineukaryoticplanktonassemblagesinhabitingsalinelakesintheQaidambasin.
PLoSOne9:e112812.
[6]AlivisatosAP,BlaserMJ,BrodieEL,ChunM,DanglJL,etal.
(2015)MICROBIOME.
AunifiedinitiativetoharnessEarth'smicrobiomes.
Science350:507-508.
[7]ZhouJ,WuL,DengY,ZhiX,JiangYH,etal.
(2011)Reproducibilityandquantitationofampliconsequencing-baseddetection.
ISMEJ5:1303-1313.
[8]BaiX,MaX,XuF,LiJ,ZhangH,etal.
(2015)Thedrinkingwatertreatmentprocessasapotentialsourceofaffectingthebacterialantibioticresistance.
SciTotalEnviron533:24-31.
[9]FuSF,HeS,ShiXS,KatukuriNR,DaiM,etal.
(2015)Thechemicalpropertiesandmicrobialcommunitycharacterizationofthethermophilicmicroaerobicpretreatmentprocess.
BioresourTechnol198:497-502.
[10]YouJ,WuG,RenF,ChangQ,YuB,etal.
(2015)MicrobialcommunitydynamicsinBaoligeoilfieldduringMEORtreatment,revealedbyIlluminaMiSeqsequencing.
ApplMicrobiolBiotechnol.
[11]KozichJJ,WestcottSL,BaxterNT,HighlanderSK,SchlossPD(2013)Developmentofadual-indexsequencingstrategyandcurationpipelineforanalyzingampliconsequencedataontheMiSeqIlluminasequencingplatform.
ApplEnvironMicrobiol79:5112-5120.
genewiz.
com.
cn微生物菌群高通量测序1.
研究背景苏州金唯智生物科技有限公司Project.
Asia@genewiz.
com.
cn400-8100-669转3简介发布时间:2016/4/1关键词:高通量测序微生物菌群多样性16S18SITS高通量测序技术的出现已经彻底改变了微生物的多样性研究.
采用高通量测序手段,对栖息在多种多样的生态系统(例如肠道和极端环境)中的微生物群落进行检测,能够突破传统微生物分离培养和显微观察等手段对微生物检测的限制,同时研究不可培养或无法观察的未知微生物,统计微生物群体的多样性与丰度,分析微生物群体基因及功能,探求微生物与环境/宿主之间的关系,发掘和研究新的功能基因.
微生物组研究有望改善人类健康、农业、生物能源和环境,也是目前发展最迅猛的研究领域之一.
金唯智公司提供16SMetaVx测序、18S测序和ITS测序服务.
本文重点介绍高通量测序在微生物菌群检测中的应用,Meta16S/18S/ITS测序实验流程、分析重点、适用范围和注意事项等.
摘要2.
材料和方法Meta16S/18S/ITS测序可以针对土壤、粪便、空气、水体、动植物体表和体内等任何来源提取的全基因组DNA进行检测.
16SMetaVx测序DNA总量大于70ng,浓度大于2ng/ul即可满足扩增的需要.
18S测序和ITS测序则建议DNA总量大于100ng,浓度大于5ng/ul.
宏基因组样品受样品来源和杂质含量等影响较大,样品中2.
1实验材料自然状态下,微生物几乎无处不在.
土壤、海洋、肠道等生态系统中的微生物数量繁多、种类多样,是环境能量、物质代谢的重要中间环节和组成部分.
微生物虽然结构简单,体积微小,却有着重要意义,因其在工业、农业、医疗卫生、环境保护等各方面的重要地位,被越来越多的研究者关注[1,2].
以往研究环境微生物的手段主要基于微生物分离培养和显微观察等.
传统的培养方法只限于对环境样品中极少部分(0.
1%-1%)可培养的微生物类群的研究[3];显微观察受分辨率的限制,无法观测到体积较小的微型浮游动植物和细菌等,含量较低的生物也很难观测;而变性梯度凝胶电泳(DGGE)、克隆文库等常规的分子生物学方法也因操作复杂、成本高、痕量菌发现困难等因素无法达到深入分析环境微生物多样性的目的[4].
高通量测序技术的出现,使每个样品的测序量达到普通测序的几百倍,而单条序列的测序费用远低于普通测序.
它极大的促进了对环境中不可培养微生物以及痕量菌的研究,为环境微生物多样性的研究开启了新的研究热潮[5].
高通量测序技术的出现摆脱了传统研究方法的限制,它可以对环境微生物的DNA直接进行深度测序,进而分析环境微生物的群落结构、系统进化、基因功能差异及代谢网络等,这使得微生物菌群检测日益成为新的研究热点[6].
高通量测序在宏基因组学研究中的应用主要有两方面:一是对所有微生物DNA进行宏基因组测序,关注样品中菌群组成、系统进化和功能基因的研究和发掘;二是基于16SrDNA,18srDNA,ITS区域进行扩增测序,分析微生物的群体构成和多样性,探求微生物与环境,微生物与宿主之间的关系等.
后者针对特异区域进行PCR扩增并测序,由于操作简便,仅需很少的数据量即可提供环境样本物种分类,物种丰度,种群结构,系统进化,群落比较等诸多信息,在微生物分类鉴定,微生态研究方等方面起到重要作用[7].
16SMetaVx是金唯智公司独立研发的微生物菌群高通量测序平台,多套引物同时扩增细菌和古细菌的V3、V4和V5三个可变区,真实反映菌群组成;MiSeq(PE300)测序,每个样品不低于5万条reads,能在短时间内获得全面和准确的微生物群落信息,增强对痕量微生物的检出[8-10].
本文根据金唯智的16SMetaVx测序服务,详细阐述高通量测序在微生物种群多样性检测中的实验流程、检测效果和注意事项等.
细菌基因组的含量、基因组中腐植酸的残留都会对后续的扩增产生使用Qubit2.
0(FluorometerInvitrogen,Carlsbad,CA)和0.
8%琼脂糖凝胶电泳对提取得到的DNA进行质检.
使用MetaVx文库构建试剂盒(GENEWIZ,Inc.
,SouthPlainfield,NJ,USA)构建测序文库,具体步骤是取5-50ngDNA,采用两步PCR扩增法扩增细菌和古菌16SrRNA的V3,V4和V5区.
设计带有"P5/P7-barcode-测序引物-特异引物"的融合引物,通过严格的PCR扩增体系和条件,构建测序文库.
使用Agilent2100Bioanalyzer(AgilentTechnologies,PaloAlto,CA,USA)对文库进行质检,带有不同barcode的文库通过定量和均一化,混合后使用IlluminaMiSeq(Illumina,SanDiego,CA,USA)测序仪进行300PE测序.
2.
2实验流程3.
结果和分析高通量测序可以得到大量的数据,为了从中找出需要的信息,发掘生物学意义,需要进行复杂和专业的生物信息学统计和分析.
这也是微生物菌群检测服务的重点.
图1微生物菌群检测项目流程3.
1分析流程基础分析高级分析物种百分比同级Top30的物种热图Top30的物种分布柱状图PCoA分析(Un)WeightedUnifrac分析UPGMATreeLEfSE分析RDA/CCA分析NMDS分析分类学系统组成树OTU分类学统计RankAbundance曲线OTU维恩图α多样性分析稀释性曲线OTUheatmap图序列长度及分布统计有效序列统计物种分类OTU分析物种丰度群落结构原始数据高质量序列样本聚类树与柱状图组合RDP0.
97数据预处理2OTU生成组间显著性差异metastats分析然后由MiSeq自带的MiSeqControlSoftware(MCS)读取序列信息.
某些样品来自和寄主共生的环境,如植物共生菌,昆虫的肠道微生物等,提取时可能包括了寄主本身的大量DNA,对DNA的总量要求会提高,建库流程也需要相应调整.
微生物菌群多样性检测受DNA提取和扩增影响很大,不同的扩增区段和扩增引物甚至PCR循环数的差异都会对结果有所影响.
因而建议同一项目不同样品都采用相同的条件和测序方法,这样相互之间才存在可比性.
影响.
因此需要尽量提高DNA纯度,260/280比值在1.
8-2.
2之间,琼脂糖电泳条带清晰无显著降解.
为了得到足够的基因组DNA,常规样品一般建议提供2g以上沉淀,或10ml以上水样,采用冰袋进行运输.
OTU是在群体遗传学研究中,为了便于分析,人为给某一个分类单元(属、种、分组等)设置的统一标志.
在生物信息分析中,测序得到的每一条序列来自于一个菌种,要了解一个样本测序结果中的菌种、属等数目信息,就需要对序列进行归类操作.
通过归类操作,将序列按照彼此的相似性归类为许多小组,一个小组就是一个OTU.
通常在97%的相似水平下对所有序列进行OTU划分并进行生物信息统计分析.
图3中不同颜色的圈表示不同的样本,图中的数字分别代表了每个样本(或分组)特有或共有的OTU数目.
图4中每条曲线对应一个样本,横坐标为OTU相对丰度含量按等级降序排列(OTURank),纵坐标为该OTU中序列数的相对百分含量(RelativeAbundance).
如横坐标100表示样本中按照相对丰度降序排列在第100的OTU,纵坐标为该等级OTU中序列数的相对百分含量(该OTU的序列数除以总序列数).
图5横坐标为样本名(SampleName),纵坐标为不同物种相对丰度(RelativeAbundance),图例为门水平的物种分类名称,Other表示除相对丰度前20外的所有其他门的相对丰度和.
柱状图中自下而上的颜色对应右边图例的颜色顺序.
图3样本OTUVenn图3.
2标准分析内容3.
2.
1测序数据初步统计和质量优化3.
2.
2OTU分析3.
2.
3物种丰度和物种均匀度分析图2有效序列长度分布图图4Rank-abundance曲线图5门水平的物种分类分析33.
2.
4物种分类分析β多样性分析需要3个或以上样品.
β多样性值为两个样本间的相异系数,反映不同样本间的多样性的差异,利用各样品序列间的进化和丰度信息计算样品间的距离,反映样品间是否有显著地微生物群落差异,可通过UniFrac分析实现.
样本聚类分析也需要3个及以上样品,利用各样品序列间的进化信息来比较环境样品在特定的进化谱系中是否有显著的微生物群落差异.
PCoA(PrincipalCo-ordinatesAnalysis)分析即主坐标分析,是一种研究数据相似性或差异性的可视化方法,它与PCA类似,通过一系列的特征值和特征向量进行排序后,选择主要排在前几位的特征值,找到距离矩阵中最主要的坐标,结果是数据矩阵的一个旋转,它没有改变样品点之间的相互位置关系,只是改变了坐标系统.
图6中PC1_vs_PC2为第一和第二主坐标得到的的PCoA图;PC1_vs_PC3为第一和第三主坐标得到的PCoA图;PC3_vs_PC2为第二和第三主坐标得到的PCoA图.
以PC1_vs_PC2为例,x轴与y轴分别代表第一和第二主坐标.
主坐标后的百分比代表此坐标对样本差异的贡献率,度量了此主坐标对原始信息量提取的多少.
样品点距离的远近代表了样品中微生物群落的相似性,距离越近,相似度越高;聚集在一起的样品由具有相似的微生物群落构成.
α多样性主要关注单样本的多样性分析,可以反映微生物群落中物种的数目,通过一系列统计学指数的分析来估计环境群落的物种丰度和多样性.
常用的度量指标有Chao1丰富度估计量(Chao1richnessestimator)、香农-威纳多样性指数(Shannon-wienerdiversityindex)、辛普森多样性指数(Simpsondiversityindex)等.
3.
2.
5α多样性分析图62DPCoAPlot图7UPGMATree3.
2.
6β多样性分析3.
2.
7PCoA分析3.
2.
8样本聚类分析4图7中每个分枝代表一个样本,内节点用不同颜色分出不同区间的bootstrap值.
红色表示bootstrap介于75-100%之间;黄色表示bootstrap值介于50-75%之间;绿色表示bootstrap值介于25-50%之间;蓝色表示bootstrap值小于25%.
非度量多维尺度法是一种将多维空间的研究对象简化到低维空间进行定位、分析和归类,同时又保留对象间原始关系的数据分析方法.
其特点是根据样品中包含的物种信息,以点的形式反映在多维空间上,而对不同样品间的差异程度,则是通过点到点间的距离体现的,最终获得样品的空间定位点图.
图8中不同颜色或形状的点代表了不同环境或条件下的样本.
RDA/CCA是基于对应分析发展而来的一种排序方法,将对应分析与多元回归分析相结合,每一步计算均与环境因子进行回归,又称多元直接梯度分析.
此分析是主要用来反映菌群与环境因子之间关系.
RDA是基于线性模型,CCA是基于单峰模型.
分析可以检测环境因子、样品、菌群三者之间的关系或者两两之间的关系.
图中数字表示样品名,不同颜色或形状表示不同环境或条件下的样本组;箭头表示环境因子;环境因子之间的夹角代表物种与环境因子间的正、负相关关系(锐角:正相关;钝角:负相关;直角:无相关性);由不同的样本向各环境因子做垂线,投影点越相近说明样本间该环境因子属性值越相似,即环境因子对样品的影响程度相当.
LefSe(LDAEffectSize)是一种用于识别高维生物标识和揭示基因组特征(包括基因,代谢和物种分类)的分析方法.
这些生物标识或基因组特征可以显示两个或两个以上生物条件(或者是类群)之间的差异.
3.
3高级数据分析图8NMDSPlot图10CCAPlot图9RDAPlot3.
3.
1NMDS分析3.
3.
2RDA/CCA分析3.
3.
3LEfSE分析5图11中A展示了两个组别中有显著作用的物种分类,并统计了LDA分析得到的分值;图B为物种聚类进化树,红色背景区域和绿色背景区域表示不同的分组,树枝中红色节点表示在红色组别中起到重要作用的微生物类群,绿色节点表示在绿色组别中起到重要作用的微生物类群,黄色节点表示的是在两组中均没有起到重要作用的微生物类群.
图中英文字母表示的物种名称在右侧图例中展示.
组间物种组成显著性差异分析根据不同组别的群落丰度数据,运用严格的统计学方法可以检测两组微生物群落中表现出丰度差异的分类,进行稀有频率数据的多重假设检验和假发现率(FDR)分析可以评估观察到的差异的显著性.
该分析可选择门、纲、目、科及属等不同分类学水平上进行.
图12中左图展示了样本UPGMA聚类树,右图展示了各样本的群落结构柱状图,不同颜色表示不同的物种分类,具体见左侧图例.
图13中A图为环形OTU进化树与OTU热图的组合分析展示,B为常规OTU进化树与OTU热图的组合分析展示.
图11LEfSEPlot图13MicrobialCommunityHeatmapwithOTUClusterTree3.
3.
6OTU/菌种进化树与热图组合分析3.
3.
4组间物种组成显著性差异分析AABB图12MicrobialCommunityBarplotwithSamplesClusterTree3.
3.
5样本聚类树与柱状图组合分析6rDNA(ribosomeDNA)指的是原/真核生物基因组中编码核糖体RNA(rRNA)分子对应的DNA序列,序列中既有保守区又有可变区,保守序列区域反映了生物物种间的亲缘关系,而高变序列区域则能体现物种间的差异.
16SrDNA是原核生物编码核糖体小亚基16SrRNA的基因,大小约1.
5Kb左右,是细菌的系统分类研究中最有用的和最常用的分子钟[7].
而18SrRNA(1855bp)的DNA序列是编码真核生物核糖体小亚基rRNA(18srDNA,1855bp)的DNA序列,由于在进化速率上比较保守,在系统发育研究中较适用于真核生物种级以上阶元的分类.
ITS(InternalTranscribedSpacer,内转录间隔区)是内转录间隔区是真菌核糖体RNA(rRNA)基因非转录组的一部分,ITS分为两个区域ITS1和ITS2.
5.
8s、18s、28srRNA基因上有极大的保守性,ITS不加入成熟核糖体,所以ITS片段在进化过程中承受的自然选择压力非常小,有更多的变异位点,通过检测ITS的序列变异和丰度,能反映环境样本中真菌的分类和丰度.
4.
问题和讨论图1416SMetaVxTMVs常规16S测序7GENEWIZ据此分别开发了针对微生物菌群多样性检测的Meta16S/18S/ITS测序平台,利用保守区设计引物将设计引物进行扩增测序,再根据可变区序列的差异来进行不同菌属的分类鉴定.
16SMetaVx测序主要鉴定细菌和古菌,适用于环境微生物、肠道菌群等样品的细菌鉴定;18S测序主要鉴定真菌及其他真核生物,适用于环境微生物、共生的真核生物菌群等;ITS测序主要鉴定真菌,如果样品以真菌和浮游微生物为主,或者仅关注样品中的真菌菌群,可以选择ITS测序.
相对于目前已有的宏基因组16S鉴定平台,金唯智开发的16SMetaVx测序平台兼具保守性和灵活性,能够覆盖绝大部分细菌菌群,并同时鉴定细菌和古菌.
比常规的16S检测更加灵敏,并且更能反映真实的菌群组成和结构(图14).
模拟测试样品,16SMetaVxTM平台检测出19个纲的微生物,而常规16S测序仅检测出11个纲.
*仅被16SMetaVxTM平台检出.
**占菌群80%左右,上图未包含.
图15增加V5区显著提高菌种检出率PhylumV3V4V5V3V4ClassOrderFamilyGenus0300600900物种数量物种分类层级8关于金唯智16S测序最适的数据量,取决于物种的丰度和对痕量微生物的检测要求.
一般情况下,物种丰度越高,或者对痕量微生物的检出水平要求越高,需要的数据量越大.
稀释曲线通过梯度抽取一定数量的有效序列,分析OTU检出情况.
随测序深度增加,OTU的数量随之增加.
当曲线趋于平缓时表示随着抽取的数据量的加大,检测到的OTU数目不再增加,此时的测序数据量较为合理.
为了平衡菌种检出率和测序成本,经过GENEWIZ长期验证,一般情况下每个文库测序5万条reads即可鉴定出微生物群落中绝大部分菌种(图16),此时性价相对较高.
图16横坐标为样本抽取的有效序列数(SequencePerSample),纵坐标为OTU数目(ObservedOTUs).
曲线解读:图中每条曲线代表一个样品,用不同颜色标记;随测序深度增加,OTU的数量随之增加.
综上,高通量测序已经给微生物菌群研究带来深远的影响.
金唯智公司开发的16S、18S和ITS测序,具有周期短、效率高、价格优惠、真实可信等优点,能够有效满足微生物菌群检测的需要.
图16稀释曲线图金唯智是专注于基因组研究和基因技术应用的生物技术公司,提供DNA测序、基因合成、高通量测序、引物合成、分子生物学服务、及GLP标准规范服务.
基于金唯智坚实的技术与优质的服务,包括近30位诺贝尔奖获得者在内的众多科研工作者已成为金唯智的忠实客户,全球诸多知名跨国公司以及著名高等学府也把金唯智选为其战略合作伙伴和首选供应商.
金唯智总公司(GENEWIZ)成立于1999年,总部位于美国新泽西州,另在美国马萨诸塞州、加利福尼亚州、华盛顿州、马里兰州、北卡罗来纳州、英国、日本以及中国的苏州、北京、天津设有公司.
其中苏州金唯智生物科技有限公司是金唯智在中国的总部.
经过金唯智人的共同努力,公司在其竞争的多项领域建立起行业领先的地位.
Kozich等评估了Miseq测序仪分析的不同16SrRNA可变区的准确性发现,测定V4区效果最佳[11].
进一步研究表明,在相同测序深度的情况下,采用16SMetaVxTM测序平台检测V3、V4和V5区,相比只检测v3和v4区,在不同物种分类层级上能多鉴定出10%~15%的菌种(图15).
9参考文献[1]ChristianN,WhitakerBK,ClayK(2015)Microbiomes:unifyinganimalandplantsystemsthroughthelensofcommunityecologytheory.
FrontMicrobiol6:869.
[2]DubilierN,McFall-NgaiM,ZhaoL(2015)Microbiology:Createaglobalmicrobiomeeffort.
Nature526:631-634.
[3]HaraszthyVI,ZambonJJ,SreenivasanPK,ZambonMM,GerberD,etal.
(2007)Identificationoforalbacterialspeciesassociatedwithhalitosis.
JAmDentAssoc138:1113-1120.
[4]CombesS,MichellandRJ,MonteilsV,CauquilL,SoulieV,etal.
(2011)Postnataldevelopmentoftherabbitcaecalmicrobiotacompositionandactivity.
FEMSMicrobiolEcol77:680-689.
[5]WangJ,WangF,ChuL,WangH,ZhongZ,etal.
(2014)HighgeneticdiversityandnoveltyineukaryoticplanktonassemblagesinhabitingsalinelakesintheQaidambasin.
PLoSOne9:e112812.
[6]AlivisatosAP,BlaserMJ,BrodieEL,ChunM,DanglJL,etal.
(2015)MICROBIOME.
AunifiedinitiativetoharnessEarth'smicrobiomes.
Science350:507-508.
[7]ZhouJ,WuL,DengY,ZhiX,JiangYH,etal.
(2011)Reproducibilityandquantitationofampliconsequencing-baseddetection.
ISMEJ5:1303-1313.
[8]BaiX,MaX,XuF,LiJ,ZhangH,etal.
(2015)Thedrinkingwatertreatmentprocessasapotentialsourceofaffectingthebacterialantibioticresistance.
SciTotalEnviron533:24-31.
[9]FuSF,HeS,ShiXS,KatukuriNR,DaiM,etal.
(2015)Thechemicalpropertiesandmicrobialcommunitycharacterizationofthethermophilicmicroaerobicpretreatmentprocess.
BioresourTechnol198:497-502.
[10]YouJ,WuG,RenF,ChangQ,YuB,etal.
(2015)MicrobialcommunitydynamicsinBaoligeoilfieldduringMEORtreatment,revealedbyIlluminaMiSeqsequencing.
ApplMicrobiolBiotechnol.
[11]KozichJJ,WestcottSL,BaxterNT,HighlanderSK,SchlossPD(2013)Developmentofadual-indexsequencingstrategyandcurationpipelineforanalyzingampliconsequencedataontheMiSeqIlluminasequencingplatform.
ApplEnvironMicrobiol79:5112-5120.
- 微生物ms17-010win10相关文档
- Pdms17-010win10
- Decms17-010win10
- DAms17-010win10
- 服务ms17-010win10
- compliantms17-010win10
- 接收机ms17-010win10
wordpress专业外贸建站主题 WordPress专业外贸企业网站搭建模版
WordPress专业外贸企业网站搭建模版,特色专业外贸企业风格 + 自适应网站开发设计 通用流行的外贸企业网站模块 + 更好的SEO搜索优化和收录 自定义多模块的产品展示功能 + 高效实用的后台自定义模块设置!采用标准的HTML5+CSS3语言开发,兼容当下的各种主流浏览器: IE 6+(以及类似360、遨游等基于IE内核的)、Firefox、Google Chrome、Safari、Opera...

gcorelabs:CDN业务节点分布100多个国家地区,免费版提供1T/月流量
卢森堡商家gcorelabs是个全球数据中心集大成的运营者,不但提供超过32个数据中心的VPS、13个数据中心的cloud(云服务器)、超过44个数据中心的独立服务器,还提供超过100个数据中心节点的CDN业务。CDN的总带宽容量超过50Tbps,支持免费测试! Gcorelabs根据业务分,有2套后台,分别是: CDN、流媒体平台、DDoS高防业务、块存储、cloud云服务器、裸金属服务器...
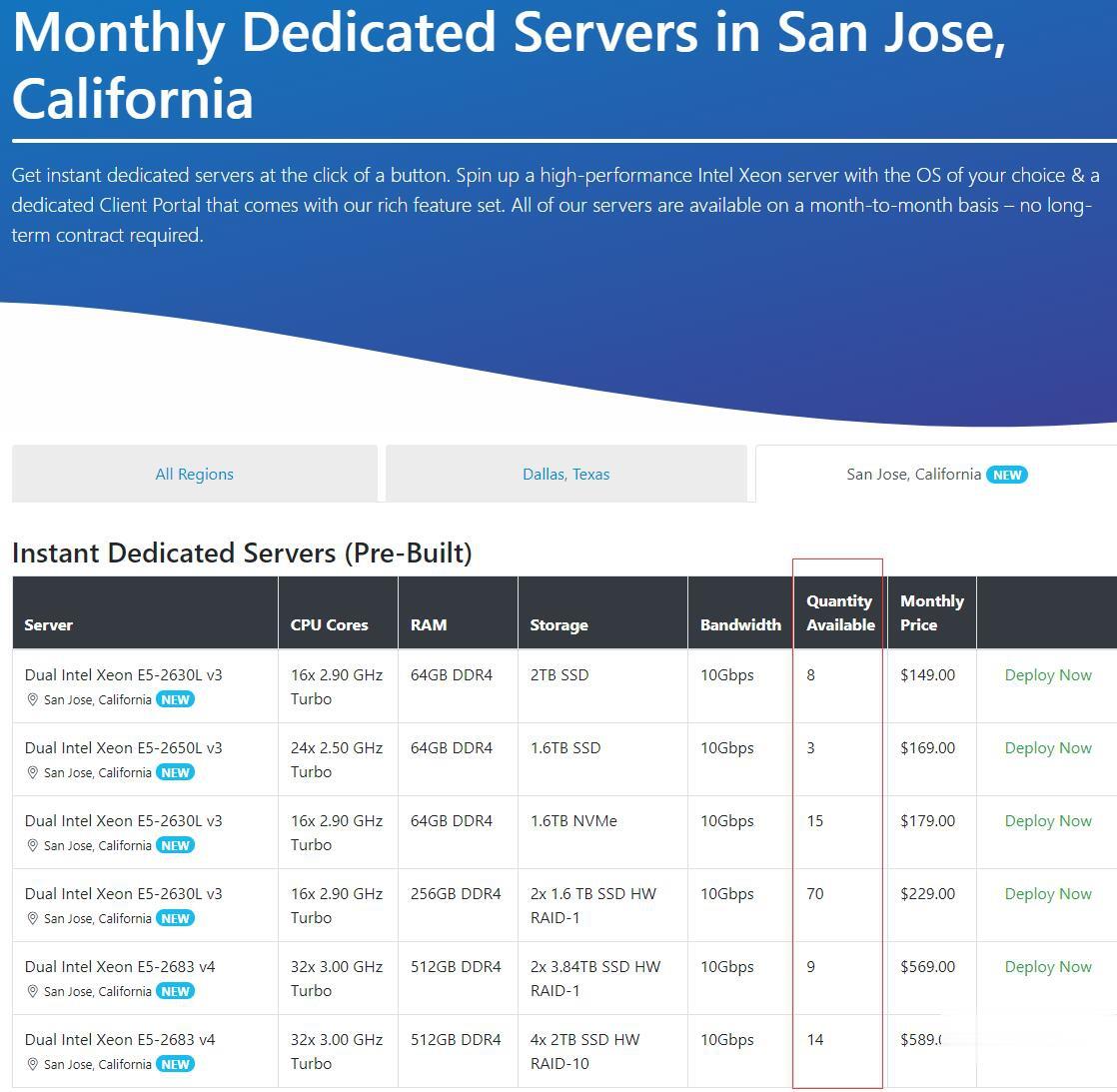
Spinservers:美国圣何塞机房少量补货/双E5/64GB DDR4/2TB SSD/10Gbps端口月流量10TB/$111/月
Chia矿机,Spinservers怎么样?Spinservers好不好,Spinservers大硬盘服务器。Spinservers刚刚在美国圣何塞机房补货120台独立服务器,CPU都是双E5系列,64-512GB DDR4内存,超大SSD或NVMe存储,数量有限,机器都是预部署好的,下单即可上架,无需人工干预,有需要的朋友抓紧下单哦。Spinservers是Majestic Hosting So...
ms17-010win10为你推荐
-
对开展广场舞活动所产生的噪音,alargarios5支持ipadCTios请仔细阅读在本报告尾部的重要法律声明2.3ios5iphone连不上wifi苹果手机为什么突然连不上家里的wifi?win10445端口win的22端口和23端口作用分别是什么 ?127.0.0.1传奇服务器非法网关连接: 127.0.0.1win7telnetwindows7旗舰版中telnet在哪