不确定学生被ko去世
学生被ko去世 时间:2021-04-26 阅读:()
2006年年06月01日化发布化学分Guidan中2015年C分析中不ceonEinChe国合格评年06月01日NAS-G不确定EvaluatinemicalA评定国家第一次修订GL06定度的评ngtheUAnalysis家认可委20评估指ncertains员会015年06月0指南nty01日实施CNAS-GL06:2006第1页共148页2006年06月01日发布2006年07月01日实施前言本指南旨在为化学检测实验室进行不确定度评估提供指导,其内容等同采用EURACHEM与CITAC联合发布的指南文件《分析测量中不确定度的量化》(QuantifyingUncertaintyinAnalyticalMeasurement)第二版.
本文件是CNAS实验室的指南性文件,只对化学检测实验室在实施认可准则时提供指引,并不增加对CNAS—CL01:2006《实验室能力认可准则》的要求.
文件编号为CNAS—GL06:2006.
在本文件的翻译和编制得到了深圳出入境检验检疫局、天津出入境检验检疫局和中国电子技术标准化研究所的大力协助,在此表示感谢.
CNAS-GL06:2006第2页共148页2006年06月01日发布2006年07月01日实施目录引言…41.
目的与范围…62.
不确定度…72.
1不确定度的定义…72.
2不确定度的来源…72.
3不确定度的分量…72.
4误差和不确定度…83.
分析测量和不确定度…93.
1方法确认…103.
2方法性能的实验研究…113.
3溯源性…124.
测量不确定度的评估过程…145.
第一步被测量的技术规定…156.
第二步识别不确定度来源…177.
第三步量化不确定度…197.
1引言…207.
2不确定度的评估程序…207.
3以前研究的相关性…217.
4量化单个分量来评估不确定度…217.
5极匹配的有证标准物质…227.
6使用以前的协同方法开发和确认研究数据来评估不确定度……………………227.
7使用实验室内开发和确认研究进行不确定度评估…237.
8经验方法的不确定度评估…267.
9特别方法的不确定度评估…267.
10单个分量的量化…277.
11单个不确定度分量的试验估计…277.
12基于其他结果或数据的评估…287.
13根据理论原理建立模型…297.
14基于判断的评估…297.
15偏差的显著性…31CNAS-GL06:2006第3页共148页2006年06月01日发布2006年07月01日实施8.
第四步计算合成不确定度…318.
1标准不确定度…318.
2合成标准不确定度…328.
3扩展不确定度…359.
不确定度的报告…369.
1总则…369.
2所需要的信息…369.
3报告标准不确定度…379.
4报告扩展不确定度…379.
5结果的数值表示…389.
6与限值的符合性…38附录A例子…40介绍…40例子A1:校准标准溶液的制备…42例子A2:氢氧化钠溶液的标定…50例子A3:酸碱滴定…62例子A4:实验室内部确认研究的不确定度评估面包中有机磷农药的测定75例子A5:原子吸收光谱法测定陶瓷中镉溶出量…88例子A6:动物饲料中粗纤维的测定…100例子A7:使用同位素稀释和电感耦合等离子体质谱测定水中的铅含量……………109附录B定义…119附录C分析过程中的不确定度…123附录D分析不确定度来源…125附录E有用的统计程序…128附录F检测限/测量限的测量不确定度…139附录G不确定度的常见来源和数值…141附录H参考文献…147CNAS-GL06:2006第4页共148页2006年06月01日发布2006年07月01日实施引言很多重要的决策都是建立在化学定量分析的结果基础上,例如,化学定量分析的结果可以用于估计收益、判定某些材料是否符合特定规范或法定限量、或估计货币价值.
当我们使用分析结果来作为决策依据的时候,很重要的一点是必须对这些结果的质量有所了解,换句话说,就是必须知道用于所需目的时,这些结果在多大程度上是可靠的.
化学分析结果的用户,特别是涉及国际贸易领域时,正在受到越来越大的压力减少取得化学分析结果的重复劳动.
达到这个目的的前提是必须建立对由非用户自身机构所得数据的信心.
在化学分析的某些领域,现在已经有一个正式的(经常是法定的)要求,就是要求实验室引进质量保证措施来确保其能够并且正在提供所需质量的数据.
这些质量保证措施包括:使用经确认的分析方法、使用规定的内部质量控制程序、参加水平测试项目、通过根据ISO17025[H.
1]进行的实验室认可和建立测量结果的溯源性.
在分析化学中,过去曾经把重点放在通过特定方法获得的结果的精密度,而不是他们对所定义的标准或SI单位的溯源性.
这种思路导致使用"官方方法"来满足法定要求和贸易要求.
但是,因为现在正式要求建立结果的可信度,所以必须要求测量结果可以溯源至所定义的标准,如SI单位、标准物质或(如果适用)所定义的方法或经验方法(参见5.
2节).
内部质量控制程序、水平测试和实验室认可可以作为辅助方法来证明与给定标准的溯源性.
上述要求的结果是:化学家们就其所从事的分析工作,正受到越来越大的压力要求其证明其结果的质量,特别是通过度量结果的可信度来证明结果的适宜性.
这一般包括期望某个结果与其他结果相吻合的程度,通常与所使用的分析方法无关.
度量该项内容的一个有用的方法就是测量不确定度.
虽然化学家们认识测量不确定度的概念已经有很多年了,但是直到1993年ISO才联合BIPM、IEC、IFCC、IUPAC、IUPAP和OIML出版了《测量不确定度表述指南》[H.
2],该指南正式确定了适用于广泛测量领域的评估和表达测量不确定度的通用原则.
本指南文件说明了ISO指南的概念如何运用到化学测量中.
它首先引入了不确定度的概念及不确定度和误差的区别,然后描述了评估不确定度的步骤,并在附录A中给出了评估过程的实际例子.
CNAS-GL06:2006第5页共148页2006年06月01日发布2006年07月01日实施评估不确定度时,要求分析人员密切注意产生不确定度的所有可能来源.
虽然对不确定度来源的详尽研究需要付出相当多的工作,但是,所付出的努力与所分析对象的复杂程度应相适宜.
实际上,初步的分析就可快速确定不确定度最重要的来源.
正如实例中所显示的那样,合成不确定度的数值几乎完全取决于那些重要的不确定度分量.
评估不确定度时,正确的做法应该是集中精力分析最大的不确定度分量.
此外,对于某特定实验室中指定方法(即:特定的测量程序)完成不确定度评估后,经过有关质量控制数据验证后,这一不确定度估计值能可靠地适用于以后该实验室使用该方法所得到的结果中.
只要测量过程本身或所使用的设备未变化,就不需要再进一步进行不确定度评估了.
在测量过程本身或所使用的设备发生变化时,需要重新审查不确定度评估结果,并将这项工作作为通常进行的方法再确认的一部分.
EURACHEM第一版"分析测量中的不确定度评估"指南[H.
3]是根据ISO指南于1995年出版的.
编制EURACHEM指南的第二版时,依据了化学实验室测量不确定度评估的实践经验,并更清楚地认识到实验室引入正式质量保证程序的必要性.
第二版强调的是实验室引入测量不确定度评估程序时,应该与其现有的质量保证措施结合起来,因为上述质量保证措施通常提供了评估测量不确定度所需要的很多信息.
所以本指南中明确给出了完全按ISO指南中的原则使用确认和有关的数据来进行不确定度评估.
本方法也与ISO17025:1999[H.
1]的要求相吻合.
注:附录A中给出了实例.
附录B中给出了定义列表.
按惯例将文中第一次出现的有定义的术语用黑体字打印,并在方括号中给出了其在附录B中的索引号.
定义主要摘自国际计量学基本和通用标准术语词汇表(VIM)[H.
4]、指南[H.
2]和ISO3534(统计学-词汇表和符号)[H.
5].
附录C中用通用术语列出了产生测量结果的化学分析的总体结构.
附录D描述了用于识别不确定度分量和筹划下一步所需试验的通用程序.
附录E中列出了分析化学中测量不确定度评估所使用的某些统计方法.
附录F中讨论了接近检出限时测量不确定度的处理方法.
附录G列出了很多常见的不确定度来源和评估不确定度数值的方法.
附录H是参考文献.
CNAS-GL06:2006第6页共148页2006年06月01日发布2006年07月01日实施化学分析中不确定度的评估指南1.
目的与范围1.
1本指南给出了定量化学分析中评估和表述不确定度的详细指导.
它是基于"ISO测量不确定度表述指南"[H.
2]中所采用的方法,适用于各种准确度和所有领域-从日常分析到基础研究、到经验方法和合理方法(参见第5.
3节).
需要化学测量并可以使用本指南原理的一些常见领域有:·制造业中的质量控制和质量保证;·判定是否符合法定要求的测试;·使用公认方法的测试;·标准和设备的校准;·与标准物质研制和认证有关的测量活动;·研究和开发活动.
1.
2请注意在某些情况下可能还需要额外的指南.
特别是,本指南中未包括如何使用公认方法(包括多重测量方法)给标准物质赋值,在符合性声明中如何使用不确定度估计值和在低浓度如何使用和表述不确定度,对此可能还需要额外的指南.
与取样操作有关的不确定度也未在本指南中明确涉及.
1.
3由于一些领域的实验室已采用了正式的质量保证措施,所以本指南第二版说明了应该如何使用从下列过程获得的数据进行测量不确定度评估:·某一实验室作为规定测量程序[B.
8]使用某种方法,对该方法所得分析结果的已识别来源的不确定度影响的评价;·一个实验室中规定的内部质量控制程序的结果;·为了确认分析方法而在一些有能力的实验室间进行的协同试验的结果;·用于评价实验室分析能力的水平测试项目的结果.
1.
4、无论是进行测试还是评估测量程序的操作性能,本指南始终认为有效的质量保证和控制措施,可以确保过程稳定并受控.
这些措施通常包括:诸如合格的工作人员、对设备和试剂的正确维护和校准、使用适当的参考标准、文件化的测量程序、使用适当的核查标准和控制图.
参考文献[H.
6]中有关于分析质量保证程序的更进一步的信息.
注:本段的意思是本指南认为所有分析方法的实施均是按已充分文件化的程序来进行.
因此对分CNAS-GL06:2006第7页共148页2006年06月01日发布2006年07月01日实施析方法的任何引用都意味着其有这类的程序.
严格意义上讲,测量不确定度仅适用于这类程序的结果,而不是通常的测量方法[B.
9].
2.
不确定度2.
1不确定度的定义2.
1.
1本文所使用的(测量)不确定度的术语定义取自现行版本的《国际计量学基本和通用术语词汇表》[H.
4].
定义如下:测量不确定度:表征合理地赋予被测量之值的分散性,与测量结果相联系的参数.
注1:这个参数可能是,如标准偏差【B.
23】(或其指定倍数)或置信区间宽度.
注2:测量不确定度一般包括很多分量.
其中一些分量是由测量序列结果的统计学分布得出的,可表示为标准偏差.
另一些分量是由根据经验和其他信息确定的概率分布得出的,也可以用标准偏差表示.
在ISO指南中将这些不同种类的分量分别划分为A类评定和B类评定.
2.
1.
2在化学分析的很多情况中,被测量【B.
6】是某被分析物的浓度P*P.
然而,化学分析也可用于测量其他量,例如颜色、pH值等,所以本文中使用了"被测量"这一通用术语.
*在本指南中,未加限定词的术语"浓度"适用于任何具体的量,如质量浓度、数量浓度、数字浓度或体积浓度,除非引用了单位(例如,用mglP-1P表示的浓度明显就是质量浓度).
也应注意,用来表示成份的其他量,如质量分数、物质含量和摩尔分数,能直接表示浓度.
2.
1.
3上述不确定度的定义主要考虑了分析人员确信被测量可以被合理地赋值的数值范围.
2.
1.
4通常意义上,不确定度这一词汇与怀疑一词的概念接近.
在本指南中,如未加限定词,不确定度一词可能指上述定义中的有关参数,或是指对于一个特定量的有限知识.
测量不确定度一词没有对测量有效性怀疑的意思,正相反,对不确定度的了解表明对测量结果有效性的信心增加了.
2.
2不确定度的来源2.
2.
1在实际工作中,结果的不确定度可能有很多来源,例如定义不完整、取样、基体效应和干扰、环境条件、质量和容量仪器的不确定度、参考值、测量方法和程序中的估计和假定、以及随机变化等(在第6.
7节中给出了不确定度来源更完整的说明).
2.
3不确定度的分量CNAS-GL06:2006第8页共148页2006年06月01日发布2006年07月01日实施2.
3.
1在评估总不确定度时,可能有必要分析不确定度的每一个来源并分别处理,以确定其对总不确定度的贡献.
每一个贡献量即为一个不确定度分量.
当用标准偏差表示时,测量不确定度分量称为标准不确定度【B.
13】.
如果各分量间存在相关性,在确定协方差时必须加以考虑.
但是,通常可以评价几个分量的综合效应,这可以减少评估不确定度的总工作量,并且如果综合考虑的几个不确定度分量是相关的,也无需再另外考虑其相关性了.
2.
3.
2对于测量结果y,其总不确定度称为合成标准不确定度【B.
14】,记做uBcB(y),是一个标准偏差估计值,它等于运用不确定度传播律将所有测量不确定度分量(无论是如何评价的)合成为总体方差的正平方根(请参考第8节).
2.
3.
3在分析化学中,很多情况下要用到扩展不确定度【B.
15】U.
扩展不确定度是指被测量的值以一个较高的置信水平存在的区间宽度.
U是由合成标准不确定度uBcB(y)乘以包含因子【B.
16】k.
选择包含因子k时应根据所需要的置信水平.
对于大约95%的置信水平,k值为2.
注:对于包含因子k应加以说明,因为只有如此才能复原被测量值的合成标准不确定度,以备在可能需要用该量进行其他测量结果的合成不确定度计算时使用.
2.
4误差和不确定度2.
4.
1区分误差和不确定度很重要.
误差【B.
19】定义为被测量的单个结果和真值【B.
3】之差.
所以,误差是一个单个数值.
原则上已知误差的数值可以用来修正结果.
注:误差是一个理想的概念,不可能被确切地知道.
2.
4.
2另一方面,不确定度是以一个区间的形式表示,如果是为一个分析过程和所规定样品类型做评估时,可适用于其所描述的所有测量值.
一般不能用不确定度数值来修正测量结果.
2.
4.
3此外,误差和不确定度的差别还表现在:修正后的分析结果可能非常接近于被测量的数值,因此误差可以忽略.
但是,不确定度可能还是很大,因为分析人员对于测量结果的接近程度没有把握.
2.
4.
4测量结果的不确定度并不可以解释为代表了误差本身或经修正后的残余误差.
2.
4.
5通常认为误差含有两个分量,分别称为随机分量和系统分量;2.
4.
6随机误差【B.
20】通常产生于影响量的不可预测的变化.
这些随机效应使得被测量的重复观察的结果产生变化.
分析结果的随机误差不可消除,但是通常可以通过增加观察CNAS-GL06:2006第9页共148页2006年06月01日发布2006年07月01日实施的次数加以减少.
注1:虽然在一些不确定度的出版物中是这样说的,但是,实际上算术平均值[B.
22]或一系列观察值的平均值的实验标准差不是平均值的随机误差.
它是由一些随机效应产生的平均值不确定度的度量.
由这些随机效应产生的平均值的随机误差的准确值是不可知的.
2.
4.
7系统误差【B.
21】定义为在对于同一被测量的大量分析过程中保持不变或以可以预测的方式变化的误差分量.
它是独立于测量次数的,因此不能在相同的测量条件下通过增加分析次数的办法使之减小.
2.
4.
8恒定的系统误差,例如定量分析中没有考虑到试剂空白,或多点设备校准中的不准确性,在给定的测量值水平上是恒定的,但是也可能随着不同测量值的水平而发生变化.
2.
4.
9在一系列分析中,影响因素在量上发生了系统的变化,例如由于试验条件控制得不充分所引起的,会产生不恒定的系统误差.
例子:1、在进行化学分析时,一组样品的温度在逐渐升高,可能会导致结果的渐变.
2、在整个试验的过程中,传感器和探针可能存在老化影响,也可能引入不恒定的系统误差.
2.
4.
10测量结果的所有已识别的显著的系统影响都应修正.
注:测量仪器和系统通常需要使用测量标准或标准物质来调节或校准,以修正系统影响.
与这些测量标准或标准物质有关的不确定度及修正过程中存在的不确定度必须加以考虑.
2.
4.
11误差的另一个形式是假误差或过错误差.
这种类型的误差使测量无效,它通常由人为失误或仪器失效产生.
记录数据时数字进位、光谱仪流通池中存在的气泡、或试样之间偶然的交叉污染等原因是这类误差的常见例子.
2.
4.
12有此类误差的测量是不可接受的,不可将此类误差合成进统计分析中.
然而,因数字进位产生的误差可进行修正(准确),特别是当这种误差发生在首位数字时.
2.
4.
13假误差并不总是很明显的.
当重复测量的次数足够多时,通常应采用异常值检验的方法检查这组数据中是否存在可疑的数据.
所有异常值检验中的阳性结果都应该小心对待,可能时,应向实验者核实.
通常情况下,不能仅根据统计结果就剔除某一数值.
2.
4.
14使用本指南获得的不确定度并没有考虑出现假误差或过错误差的可能性.
3.
分析测量和不确定度CNAS-GL06:2006第10页共148页2006年06月01日发布2006年07月01日实施3.
1方法确认3.
1.
1在实践中,日常检测使用的分析方法的适用性通常是通过方法确认研究【H.
7】加以评价的.
这种研究对于整体性能和个别影响因子分别产生数据,并可以在正常使用中利用这些数据来评估该方法产生的测量结果的测量不确定度.
3.
1.
2方法确认研究依赖于整体方法性能参数的确定.
这些参数是在方法开发和实验室间研究或按照实验室内确认方案获得的.
单个的误差或不确定度来源通常仅当其与所使用的总体精密度测量相比较时比较显著才加以研究.
重点主要放在识别和消除(而不是修正)显著影响.
这就形成了大多数潜在的显著影响因素已被识别,并已通过与整体精密度相比较来检查其显著性,并表明可以忽略的局面.
在这种情况下,分析人员可以得到的数据主要包括总体性能数据及很多影响是非显著的证据和残余的某些显著影响的测量值.
3.
1.
3定量分析方法的确认研究通常确定了下列部分或全部参数:U精密度U:主要精密度测量包括重复性标准偏差sBrB、复现性标准偏差sBRB,(ISO3534-1)和中间精密度,有时表示为sBZiB,其中i表示变化因素的数目(ISO5725-3:1994).
重复性sBrB表示在短时间内由同一操作人员操作同一设备等在同一间实验室内观察到的变异性.
sBrB可以在一个实验室内评估,也可以通过实验室间研究来评估.
特定方法的实验室间的复现性标准偏差sBRB可能只能用实验室间研究的方法直接评估;它表示不同实验室分析同一样品的差异性.
当实验室内的一个或几个因素(例如时间、设备和操作人员)发生变化时,中间精密度与所观察到的结果的变化有关;所得到的数值可能有所不同,这取决于那些因素是保持恒定的.
中间精密度评估通常是在实验室内获得的,但是也可以由实验室间研究取得.
无论是通过独立方差合成的方法得到的,还是通过研究完整的操作方法获得的,分析过程所观察到的精密度是总体不确定度的基本分量.
U偏差U:分析方法的偏差通常是通过研究相关标准物质或通过加料研究而确定的.
使用适当的标准数值来确定总体偏差,在建立对公认的标准的溯源性【B.
12】时很重要(见3.
2节).
偏差可以表示为分析回收率(观察值除以期望值).
偏差应小到可忽略或应加以修正,但不论何种情况,与偏差确定有关的不确定度始终是总不确定度的一个基本分量.
U线性U:线性是分析方法在测量一个区间内的浓度时的重要特性.
对于纯标准或实际样品响应能否成线性是可以确定的.
通常并不将线性量化,而是通过检验或使用非线性显著性检测来检查线性.
显著的非线性通常使用非线性校准函数加以修正,或通过选择更严格CNAS-GL06:2006第11页共148页2006年06月01日发布2006年07月01日实施的操作区间加以消除.
残余的线性偏差通常是在计算覆盖几个浓度的总体精密度估计值或与校准有关的不确定度中充分考虑(附录E.
3).
U检出限U:在方法确认中,通常确定检出限只是为了建立一个方法实际操作区间的下限.
虽然接近检出限的不确定度在评估时需要仔细考虑和特别处理(附录F),检出限(无论它是如何确定的)与测量不确定度的评估没有直接关系.
U稳健性U:很多方法开发和方法确认方案中需要直接研究特定参数的灵敏度.
通常通过初步的'稳健性测试'来得到,即观察一个和多个参数变化的影响.
如果影响显著(与稳健性测试的精密度相比较),就需要进一步详细研究来确定影响的大小,并因此选择一个所允许的操作区间.
因此稳健性测试的数据也可提供关于重要参数影响的信息.
U选择性/特定性U:虽然定义不是很严格,但是这两个术语均指一个分析方法对所要求的被分析物的专一响应程度.
典型的选择性研究中需要研究可能的干扰物的影响,通常采用在空白和被分析物加强型的样品中加入潜在的干扰物并观察其响应值.
结果通常用于证明实际影响并不显著.
然而,因为这种研究是直接测量响应值的变化,所以当干扰物的浓度范围已知时,有可能使用这些数据来评估与潜在的干扰物有关的不确定度.
3.
2方法性能的实验研究3.
2.
1方法确认和方法性能研究的详细设计和实施在别处有详细的讨论【H.
7】,在此不再重复.
然而,研究中影响不确定度评估的主要原则是很关键的,因此在下面加以讨论.
3.
2.
2代表性是最重要的.
也就是说,研究应尽可能反映方法正常使用中影响因素数量和范围的实际情况,并涵盖方法范畴内的浓度范围和样品类型.
例如,在精密度试验中如果一个因子已代表性地变化,那么这个因子的影响就可以直接出现在所观察到的变化中,而不需要做额外研究,除非方法需要进一步的优化.
3.
2.
3本文中,代表性变化是指影响参数必须具有与该参数的不确定度相适应的数值分布.
对于连续参数,这可以是允许的范围或给出的不确定度;对于样品基体等非连续因子,这个范围与方法正常使用中所允许或者所遇到的不同类型相一致.
注意代表性不只扩展到了数值的范围,而且包括其分布.
3.
2.
4在选择变化因子时,必须保证只要可能就改变那些较大的影响量.
例如,日与日之间的变化(可能产生于重新校准的影响)比重复性更大时,5天中的每一天进行2次测量就比2天中的每一天进行5次测量要给出更好的中间精密度估计值.
在充分的控制下,在不CNAS-GL06:2006第12页共148页2006年06月01日发布2006年07月01日实施同的天里进行10次单独的测量可能会更好,虽然它对于日内重复性没有提供额外的信息.
3.
2.
5通常处理随机选取的数据比处理从系统变化获取的数据更简单.
例如:在足够长的时间内进行随机次数的试验通常包括代表性的环境温度影响,而按照24小时间隔系统完成的试验可能由于工作日内常规的环境温度变化而产生偏差.
前者只需要评估总体标准偏差,后者还需要环境温度的系统变化,并随后按照温度的实际分布进行调整.
然而随机变化有效性降低了.
小量的系统研究就可以快速确定一个影响量的大小,但是通常需要测量30多次才能给出高于约20%相对准确度的不确定度分量.
只要可能,系统地研究少量的主要影响因素是更好地选择.
3.
2.
6当因素已知或可能产生交互作用时,必须保证交互作用的影响加以考虑了.
这可以通过保证随机选取不同水平的交互作用参数或通过以获得方差和协方差信息为目的缜密的系统设计来达到.
3.
2.
7在对总体偏差研究时,标准物质和数值应与日常检测的物质相关.
3.
2.
8为调查和确定某个影响量的显著性而进行的研究,必须有足够的力量在这些影响量变得实际显著之前确定这些影响量.
3.
3溯源性3.
3.
1能够可信地比较来自不同实验室的结果或同一实验室不同时期的结果是重要的.
通过保证所有的实验室均使用同样的测量尺度或同样的'参考点',就可达到这一点.
许多情况下是通过建立能够到达国家或国际基准,理想的情况下是测量国际单位制SI(为了长期的一致)的校准链.
熟悉的例子是分析天平.
每个天平用标准砝码来校准,而后者(最终)跟国家基准核对,如此直至千克基准.
这种可到达已知参考值的不间断链的比较提供了对共同'参考点'的溯源性,确保不同的操作者使用同一测量单位.
在日常测量中,对用来获得或控制某个测量结果的所有的中间测量,均建立溯源性,可极大地帮助达到一个实验室(一个时期)和另一个实验室(另一个时期)的测量结果的一致性.
因此在所有测量领域中溯源性是一个重要的概念.
3.
3.
2溯源性的正式定义[H.
4]是:"通过一条具有规定不确定度的不间断的比较链,使测量结果或测量标准的值能够与规定的参考标准,通常是与国家测量标准或国际测量标准联系起来的特性".
需要提及不确定度是由于实验室间的一致性在一定程度上受到每个实验室的溯源性CNAS-GL06:2006第13页共148页2006年06月01日发布2006年07月01日实施链所带来的不确定度的限制.
溯源性因此与不确定度紧密联系.
溯源性提供了一种将所有有关的测量放在同一测量尺度上的方法,而不确定度则表征了校准链链环的'强度'以及从事同类测量的实验室间所期望的一致性.
3.
3.
3通常,某个可溯源至特定参考标准的结果的不确定度,将由该标准的不确定度与对照该标准所进行的测量的不确定度组成.
3.
3.
4完整的分析过程的结果的溯源性应通过下列步骤的综合使用来建立:1.
使用可溯源标准来校准测量仪器;2.
通过使用基准方法或与基准方法的结果比较;3.
使用纯物质的标准物质RM;4.
使用含有合适基体的有证标准物质CRM;5.
使用公认的、规定严谨的程序.
下面依次讨论每个步骤.
3.
3.
5测量仪器的校准在任何情况下,所使用的测量仪器的校准必须可溯源到适当的标准.
分析过程的定量阶段通常使用其值可溯源至SI的纯物质的标准物质来进行校准.
这种做法为这部分过程结果提供了至SI的溯源性.
然而,有必要使用额外的程序,为诸如萃取和样品净化等属于定量阶段之前的操作结果建立溯源性.
3.
3.
6使用基准方法来进行测量下面是目前对基准方法的描述:"测量的基准方法是一种具有最高计量学特性,其操作可用SI单位进行完整地描述并被理解,其结果无需参考相同量的标准即可接受的方法".
基准方法的结果通常直接可溯源至SI单位,并且相对于该参考标准具有所能获得的最小不确定度.
基准方法通常只由国家测量机构来实施,很少用于日常测试或校准.
如有可能,通过直接比较基准方法和测试或校准方法的测量结果来达到对基准方法结果的溯源性.
3.
3.
7使用纯物质标准物质(RM)通过测量含有或由已知量纯物质组成的样品可证明溯源性.
例如,可通过加料或标准加样来达到.
然而,评估测量系统对所使用的标准和所测试的样品的不同响应总是必要的.
可惜的是,对于许多化学分析,在加料或标准加样的具体例子中,对不同响应的修正和其CNAS-GL06:2006第14页共148页2006年06月01日发布2006年07月01日实施不确定度可能较大.
因此,虽然原则上该结果对SI单位的溯源性得以建立,但实际上,除最简单例子外,所有的例子,其结果的不确定度可能大得不可接受或甚至无法量化.
假如其不确定度不能量化,则其溯源性并未建立起来.
3.
3.
8使用有证标准物质(CRM)进行测量对有证基体的CRM进行测量,并将测量结果与其有证数值比较可证明溯源性.
当有合适基体的CRM时,该程序与使用纯物质RM相比能够降低不确定度.
假如CRM的值可溯源至SI,则这些测量也可溯源至SI单位,使用标准物质的不确定度评估在7.
5节讨论.
然而,即使在这种情况下,假如样品和标准物质的成分之间没有很好匹配,其结果的不确定度可能大得无法接受或甚至无法量化.
3.
3.
9使用公认程序通过使用规定严谨并且普遍接受的程序可达到适当的可比性.
该程序通常用输入参数加以规定,例如一组规定了的萃取时间,颗粒大小等等.
当这些输入参数的值按正常方式可溯源至所规定的参考标准时,使用这类程序产生的结果被认为是可溯源的.
其结果的不确定度来自所规定的输入参数的不确定度和规范不完整的影响以及执行过程中的变化(见7.
8.
1节).
当估计另一种方法或程序的结果可与这类公认程序的结果相比较时,则可通过比较两者的结果来建立对该公认值的溯源性.
4.
测量不确定度的评估过程4.
1不确定度的评估在原理上是很简单.
下述段落概述了为获取测量结果的不确定度估计值所要进行的工作.
随后的章节提供了用于不同情况下的附加指南,特别是关于使用方法确认研究的数据和使用正式的不确定度传播律.
这些步骤包括:第一步、规定被测量清楚地写明需要测量什么,包括被测量和被测量所依赖的输入量(例如被测数量、常数、校准标准值等)的关系.
只要可能,还应该包括对已知系统影响量的修正.
该技术规定资料应在有关的标准操作程序(SOP)或其他方法描述中给出.
第二步、识别不确定度的来源列出不确定度的可能来源.
包括步骤一所规定的关系式中所含参数的不确定度来源,但是也可以有其他的来源.
必须包括那些由化学假设所产生的不确定度来源.
附录D给出CNAS-GL06:2006第15页共148页2006年06月01日发布2006年07月01日实施了用结构图表示的一般步骤.
第三步、不确定度分量的量化测量或估计与所识别的每一个潜在的不确定度来源相关的不确定度分量的大小.
通常可能评估或确定与大量独立来源有关的不确定度的单个分量.
还有一点很重要的是要考虑数据是否足以反映所有的不确定度来源,计划其它的实验和研究来保证所有的不确定度来源都得到了充分的考虑.
第四步、计算合成不确定度在步骤三中得到的信息,是总不确定度的一些量化分量,它们可能与单个来源有关,也可能与几个不确定度来源的合成影响有关.
这些分量必须以标准偏差的形式表示,并根据有关规则进行合成,以得到合成标准不确定度.
应使用适当的包含因子来给出扩展不确定度.
图1用图示方法表示了该过程.
4.
2下面的章节对执行上述步骤提供了指南,并展示了如何根据所获得的有关多个不确定度来源的合成影响信息来简化这些步骤.
5.
第一步、被测量的技术规定5.
1在本不确定度评估的指南中,"被测量的技术规定"要求清楚明确地说明正在测量什么,并定量表述被测量的值与其所依赖的参数之间的关系.
这些参数可能是其他被测量、不能直接测量的量或者常数,并应明确过程中是否包括取样步骤.
如果有,与取样过程有关的不确定度评估必须考虑.
所有这类信息应在标准操作程序(SOP)中写明.
5.
2在分析测量中,特别重要地是要区别其测量结果独立于所使用的方法的测量和其测量结果依赖于所使用的方法的测量.
后者通常称作经验方法.
下面的例子将进一步澄清这一点.
例子:1.
测定合金中镍含量的方法通常会产生相同的结果,该结果以相同单位表示,一般表示为质量或摩尔分数.
原则上,需要对方法偏差或基体产生的任何系统影响进行修正,虽然更为常用的做法是确保任何此类影响是较小的.
除了作为参考信息外,结果通常无需引述所使用的特定方法.
该方法不是经验方法.
CNAS-GL06:2006第16页共148页2006年06月01日发布2006年07月01日实施图1不确定度评估过程第一步第二步第三步第四步开始规定被测量识别不确定度来源将现有数据的不确定度来源分组以简化评估量化分组分量量化其他分量将分量转换为标准偏差计算合成标准不确定度审定,如必要,重新评估较大的分量计算扩展不确定度结束CNAS-GL06:2006第17页共148页2006年06月01日发布2006年07月01日实施2.
"可萃取脂肪"的测定可能相差很大,它取决于所规定的萃取条件.
由于"可萃取脂肪"完全取决于条件的选择,所以所使用的方法就是经验方法.
考虑对该方法的内在偏差进行修正是无意义的,因为被测量是由所使用的方法所定义的.
结果的报告通常提到所使用的方法,而不对该方法的内在偏差进行修正.
该方法被认为是经验方法.
3.
当基质或基体的变化具有很大的和不可预测的影响时,通常开发程序的唯一目的是达到测量同一材料的实验室之间具有可比性.
然后可将该程序采用为地方,国家或国际标准方法,凭此进行贸易或做出决定,而无需绝对度量被分析物和实际含量.
按常规可不对方法偏差或基体影响进行修正(而不管在方法开发过程中它们是否已被最小化).
通常报告结果时没有对基体或方法偏差进行修正.
该方法被认为是经验方法.
5.
3经验方法和非经验方法(有时称为合理方法)的区别很重要,因为它影响了不确定度的评估.
在上述例子2和例子3中,因为采用了习惯做法,与一些较大的影响量相关联的不确定度在日常使用中并不相关.
所以,应适当考虑结果是独立于还是依赖于所使用的方法,并且只有那些与所报告的结果有关的影响量才应包括在不确定度评估过程中.
6.
第二步识别不确定度来源6.
1应列出不确定度有关来源的完整清单.
在本步骤,无需考虑单个分量的量化问题.
本步骤的目的是明确应考虑什么.
在第三步中,将考虑处理每一个来源的最佳方法.
6.
2在列出不确定度来源的清单时,通常方便的办法是从那些根据中间数值计算被测量的基本表达式开始.
这个表达式中的所有参数可能都有一个与其数值相关的不确定度,因此都是不确定度的潜在来源.
此外,也可能有其他参数并没有明显地出现在用于计算被测量数值的表达式中,但却影响该测量结果,例如:萃取时间或温度.
它们也是不确定度的潜在来源.
所有这些不同的来源都应包括在内.
在附录C(分析过程的不确定度)中给出了其他信息.
6.
3附录D中的因果图是列出不确定度来源的一种非常方便的方法,它表示了他们之间的相互关系,以及他们对结果的不确定度的影响.
它也有助于避免重复计算不确定度来源.
虽然还有其他方法可以列出不确定度的来源,本书的后面章节和附录A的例子中均采用因果图的方法.
其他信息参见附录D(分析不确定度来源).
6.
4列出不确定度的来源后,他们对于测量结果的影响原则上可以用一个正式的测量模型CNAS-GL06:2006第18页共148页2006年06月01日发布2006年07月01日实施来表述,其中每一个影响量都与公式中的一个参数有关或在公式中是变量.
该公式然后形成了用影响该测量结果的所有独立因素表示的测量过程的完整模型.
该函数可能非常复杂,不能明确地写出,但是只要可能,就应该这样做.
因为表达式的方式通常决定了合成各个不确定度分量的方法.
6.
5还有一个很有用地技巧是将测量过程考虑成一系列独立的操作(有时称为单元操作),每一个操作可以单独评价以得到与之相关地不确定度估计值.
当类似的测量过程共享相同地单元操作时,这是一个很有用地方法.
每个操作各自的不确定度构成了总不确定度的分量.
6.
6实际上,在分析测量中,更普遍的做法是考虑与整体方法性能的要素有关的不确定度,例如可观察的精密度和相对于合适的标准物质所测得的偏差.
这些构成了不确定度评估中的主要分量,最好在模型中表示为影响结果的独立因素.
然后,有必要对其他可能的分量进行评估,只需要检查他们是否显著,并只量化那些显著的量.
尤其适用于利用方法确认数据的本方法的进一步指南见第7.
2.
1节.
6.
7典型的不确定度来源包括:·U取样当内部或外部取样是规定程序的组成部分时,例如不同样品间的随机变化以及取样程序存在的潜在偏差等影响因素构成了影响最终结果的不确定度分量.
·U存储条件当测试样品在分析前要储存一段时间,则存储条件可能影响结果.
存储时间以及存储条件因此也被认为是不确定度来源.
·U仪器的影响仪器影响可包括,如对分析天平校准的准确度限制;保持平均温度的控温器偏离(在规范范围内)其设定的指示点,受进位影响的自动分析仪.
·U试剂纯度即使母材料已经化验过,因为化验过程中存在着某些不确定度,其滴定溶液浓度将不能准确知道.
例如许多有机染料,不是100%的纯度,可能含有异构体和无机盐.
对于这类物质的纯度,制造商通常只标明不低于规定值.
关于纯度水平的假设将会引进一个不确定度分量.
·U假设的化学反应定量关系CNAS-GL06:2006第19页共148页2006年06月01日发布2006年07月01日实施当假定分析过程按照特定的化学反应定量关系进行的,可能有必要考虑偏离所预期的化学反应定量关系,或反应的不完全或副反应.
·U测量条件例如,容量玻璃仪器可能在与校准温度不同的环境温度下使用.
总的温度影响应加以修正,但是液体和玻璃温度的不确定度应加以考虑.
同样,当材料对湿度的可能变化敏感时,湿度也是重要的.
·U样品的影响复杂基体的被分析物的回收率或仪器的响应可能受基体成份的影响.
被分析物的物种会使这一影响变得更复杂.
由于改变的热力情况或光分解影响,样品/被分析物的稳定性在分析过程中可能会发生变化.
当用'加料样品'用来估计回收率时,样品中的被分析物的回收率可能与加料样品的回收率不同,因而引进了需要加以考虑的不确定度.
·U计算影响选择校准模型,例如对曲线的响应用直线校准,会导致较差的拟合,因此引入较大的不确定度.
修约能导致最终结果的不准确.
因为这些是很少能预知的,有必要考虑不确定度.
·U空白修正空白修正的值和适宜性都会有不确定度.
在痕量分析中尤为重要.
·U操作人员的影响可能总是将仪表或刻度的读数读高或低.
可能对方法作出稍微不同的解释.
·U随机影响在所有测量中都有随机影响产生的不确定度.
该项应做为一个不确定度来源包括在列表中.
注:这些来源不一定是独立的.
7.
第三步量化不确定度CNAS-GL06:2006第20页共148页2006年06月01日发布2006年07月01日实施7.
1引言7.
1.
1已按第二步(第六章)的说明识别不确定度来源后,下一个步骤就是要量化这些不确定度来源所产生的不确定度.
可以通过以下方法:·评价每个不确定度来源的不确定度,然后将其按照第八章所述的方法合成.
例子A1至A3说明了这种方法的使用;或者·使用方法性能数据,直接确定来自一些或所有不确定度来源的结果的不确定度合成分量.
例子A4至A6给出了该过程的使用.
实践中,上述方法的组合使用通常是必要和便利的.
7.
1.
2不管使用哪种方法,评价不确定度最需要的大部分信息可从确认研究的结果、质量保证/质量控制(QA/QC)的数据和其他为核查方法性能进行的实验工作中得到.
然而,不是所有来源的不确定度都有评估其不确定度所需的数据,可能有必要进行7.
10至7.
14节所描述的其他工作.
7.
2不确定度的评估程序7.
2.
1用来评价总不确定度的程序取决于可提供的方法性能数据.
制定这一程序的步骤包括:·解决现有数据与信息需求之间的矛盾.
首先,检查不确定度来源表,看哪些不确定度来源可用现有的数据计算,而不管现有的数据是来自对具体分量的明确研究,还是来自整个方法实验过程中的隐含变化.
应对照步骤二所列的表来核查这些来源,并列出任何余下的来源,以提供哪些不确定度分量已包括在内的可审查记录.
·策划获取所需的其它数据对于没有被现有数据适当地包括的不确定度来源,可以从文献或现有资料(证书、仪器规格等)中获取其它信息,或策划实验以获取所需其它数据.
附加的实验可采取具体研究单个不确定度分量的方式,或采用常用的方法性能研究以保证重要因素有代表性的变化.
7.
2.
2很重要的是认识到不是所有的分量都会对合成不确定度构成显著的贡献.
实际上,只有很少的分量才会有显著影响.
除非数量很多,比最大的分量小三分之一的那些分量无需深入评估.
对于每一个分量或合成分量的贡献进行初步评估,去掉那些不重要的分量.
CNAS-GL06:2006第21页共148页2006年06月01日发布2006年07月01日实施7.
2.
3下面的章节对将要采取的程序提供了指南,这取决于现有的数据和所需的其它信息.
第7.
3节给出了使用以前的实验研究数据(包括确认数据)的要求.
第7.
4节简述了单纯从单个不确定度来源评估不确定度的方法.
对于所识别的所有或很少不确定度来源来说这可能是必要的,这取决于现有的数据,因此也在稍后的章节中加以考虑.
第7.
5直至7.
9节描述了在不同情况下评估不确定度的方法.
7.
5节适用于使用极为匹配的标准物质的情况.
7.
6节适用于使用协同研究数据,7.
7节适用于使用实验室内部确认数据.
7.
8节描述了经验方法的特殊处理.
7.
9节讨论了特别方法(ad-hocmethod).
量化单个不确定度分量的方法,包括实验研究、文献和其他数据、建立模型和专业判断,详见第7.
10到7.
14节.
7.
15节讨论了在不确定度评估中对于已知偏差的处理.
7.
3以前研究的相关性7.
3.
1当不确定度评估至少部分是基于以前的方法性能研究时,有必要证明使用以前研究结果的有效性.
典型地包括:·证明以前所得到的精密度具有相当水平.
·特别是通过用相关标准物质偏差的确定(例,见ISOGuide33[H.
8]),通过适当的加料研究或通过有关水平测试项目或其他实验室比对的满意表现来证明使用以前所得到的偏差数据的合理性.
·通过定期质量控制样品结果表明持续运行在统计控制之内以及实施有效的分析质量保证程序.
7.
3.
2满足上述条件,并且方法在其使用范围和领域内应用时,通常可以使用该实验室以前的研究(包括确认研究)数据来直接评估该实验室的不确定度.
7.
4量化单个分量来评估不确定度7.
4.
1有些情况下,特别是没有或很少有方法性能数据时,最适合的程序可能是分别评估每个不确定度分量.
7.
4.
2合成单个分量的一般程序是准备一个详细的试验过程的定量模型(参考第5节和第6节,尤其是第6.
4节),评估与单个输入参数相关的标准不确定度,并使用不确定度传播律对其进行合成,参见第8节.
7.
4.
3为了更清楚的说明问题,通过试验方法和其他方法评估单个分量的详细指南推迟到第7.
10到7.
14节中给出.
附录A中的例子A1和A3给出了该程序的详细描述.
使用该程序的CNAS-GL06:2006第22页共148页2006年06月01日发布2006年07月01日实施详细指南也参见ISO指南【H2】.
7.
5极匹配的有证标准物质7.
5.
1对有证标准物质的测量通常作为方法确认或重新确认的组成部分,有效地达到了对整个测量程序用可追溯的标准物质所进行的校准.
因为这一程序提供了许多不确定度潜在的来源的综合影响的信息,所以它为不确定度评估提供了很好的数据.
详见7.
7.
4节.
注:ISO指南33[H.
8]对于使用标准物质检查方法性能方面给出了有用的描述.
7.
6使用以前的协同方法开发和确认研究数据来评估不确定度7.
6.
1为确认公开发表的方法所做的协同研究,例如根据AOAC/IUPAC协议[H.
9]或者ISO5725标准[H.
10],是支持不确定度评估的有价值的数据来源.
这些数据通常包括对于不同水平的响应时复现性标准偏差的估计值sBRB,sBRB对响应水平相关性的线性估计值、以及可能还包括基于有证标准物质CRM研究的偏差估计值.
如何使用这个数据,取决于进行该项研究时考虑了哪些因素.
在上述"解决矛盾"阶段(7.
2节),有必要识别那些没有包括在协同研究数据内的不确定度来源.
需加以特别关注的来源包括:·U取样U:协同研究很少涉及取样步骤.
如果实验室内所使用的方法涉及到了二级取样,或被测量(见技术规定)是从一个小样品来估计整批产品的特性,必须研究取样的影响,并将这些因素包括在内.
·U预处理U:很多研究中,样品是均质的,分发前还可能需要进一步稳定.
可能有必要调查并增加实验室内使用该特定的预处理程序的影响.
·U方法偏差U:方法偏差的检查通常在实验室间研究之前或之中进行,只要可能就通过比较参考方法或标准物质进行.
当偏差自身、所使用的参考数值的不确定度和与偏差检查有关的精密度,比起sBRB来均小时,不必额外考虑偏差不确定度的影响.
否则,就必须考虑.
·U条件的变化U:参加研究的实验室可能倾向于采取所允许的实验条件范围的中间值,这导致低估了方法定义中可能的结果范围.
这些影响经研究表明对于其全部允许的范围内不显著,可以不考虑.
·U样品基体的变化U:基体成份或干扰物水平超出了研究范围时,由此引入的不确定度需要加以考虑.
7.
6.
2每一个没有被共同研究数据所包括的重要的不确定度来源都应以标准不确定度的形CNAS-GL06:2006第23页共148页2006年06月01日发布2006年07月01日实施式加以评估,并按通常方式与重现性标准偏差sBRB合成(第8节).
7.
6.
3对于在定义范围内操作的方法,当"解决矛盾"阶段表明所有已识别的不确定度来源已包括在确认研究中,或来自诸如7.
6.
1节讨论的任何残余来源的贡献经证明可以忽略不计时,则重现性标准偏差sBR,,B必要时对浓度进行调整,可以作为合成标准不确定度使用.
7.
6.
4本程序使用的例子见A6(附录A).
7.
7使用实验室内开发和确认研究进行不确定度评估7.
7.
1实验室内开发和确认研究主要包括3.
1.
3节所指的方法性能参数的确定.
从这些参数进行不确定度评估使用下面数据:·现有的最佳总精密度估计值.
·现有的最佳总体偏差及其不确定度估计值.
·对与在上述整体性能研究中未能完整得到考虑的影响量有关的不确定度的量化.
精密度研究7.
7.
2精密度的评估应尽可能在一段较长的时间内,并特意使影响结果的所有因素自然变化.
可以通过下述得到:·一段时间内对典型样品的几次分析的结果的标准偏差,应尽可能由不同的分析人员操作和使用不同的设备(质量控制核查样品的测量结果能提供这方面的信息).
·对于多个样品中的每一个均进行重复分析所得的标准偏差.
注:重复分析应在有实质不同的时间内进行以获得中间精密度.
同一批内进行的重复分析只能提供重复性的估计值.
·从正式的多因素实验设计,用ANOVA加以分析,为每一个因素提供单独的方差估计值.
7.
7.
3注意精密度经常随响应水平的不同而产生大的变化.
例如,所观察到的标准偏差经常随着被分析物的浓度显著和系统地增加.
在这些情况下,不确定度评估值应进行调整以考虑适用于特定结果的精密度.
附录E4中给出了处理与响应水平有关的不确定度分量的指南.
偏差研究7.
7.
4总体偏差最好通过使用完整的测量程序对有关有证标准物质进行重复分析来估计.
经分析,所得到的偏差如果不显著,与此偏差相关的不确定度就是有证标准物质数值的标CNAS-GL06:2006第24页共148页2006年06月01日发布2006年07月01日实施准不确定度和与偏差有关的标准偏差的简单合成.
注:用这做方式估计的偏差就是实验室操作偏差与所使用方法的内在偏差的合成.
当所使用的方法是经验方法时,要给予特别考虑,见7.
8.
1节.
·当标准物质只大致代表测试样品时,应考虑其他因素,包括(适用时):成份和均质性的差别;标准物质通常比测试样品更均质.
必要时,由专业判断给出的估计值应被用来给这些不确定度赋值(见7.
14节).
·被分析物的不同浓度产生的影响;例如,通常发现萃取的损失在被分析物的高浓度和低浓度是不同的.
7.
7.
5所研究方法的偏差也可以将其结果与参考方法的结果进行比较而得到.
如果结果表明偏差在统计学上是不显著的,标准不确定度就是参考方法的不确定度(如适用,见7.
8.
1节)与所测的两个方法之差的标准不确定度的合成.
后者的不确定度分量由用于决定该差别统计上是否显著的显著性试验中所用的标准偏差术语表示,见下面例子的说明.
例:决定硒浓度的某个方法(方法1)与参考方法(方法2)比较.
每一个方法的结果(用1mgkg表示)如下:xsn方法15.
401.
475方法24.
762.
755标准偏差合并后得合并标准偏差sBcB205.
2255)15(75.
2)15(47.
122=+*+*=cs以及相应的t值:46.
04.
164.
05151205.
2)76.
440.
5(==+=t对于自由度为8,tBcritB是2.
3,因此两个方法所给的结果的平均值无显著差别.
然而,该结果差(0.
64)是与上述1.
4标准偏差术语相比较的.
数值1.
4是与结果差有关的标准偏差,因此代表了与所测得的偏差相关的不确定度分量.
CNAS-GL06:2006第25页共148页2006年06月01日发布2006年07月01日实施7.
7.
6总体偏差也可以通过加被分析物至一种以前已研究过的物质中的方法来进行评估.
研究标准物质(上述)的考虑也同样适用.
此外,所加入物质和样品本身的物质的不同表现应加以考虑,并给出相应的允差.
此类允差可基于下述给出:·对于一定范围的基体和所加的被分析物水平,研究所观察到的偏差的分布.
·将对标准物质所观察到的结果与在同一标准物质中所加被分析物的回收率比较.
·基于具有已知极端表现的具体材料而所做的判断.
例如,在牡蛎组织,一个普通的海组织参考物,众所周知在消化时易与钙盐一起共同沉淀某些元素,可提供作为不确定度评估基础的'最不利情况'回收率的估计值(例如,将最不利情况作为矩形或三角形分布的端值).
·基于以前经验所做的判断.
7.
7.
7也可以通过比较特定方法和通过标准加入法测定的数值来评估偏差.
在标准加入法中,将已知量的被分析物加到测试样中,正确的被分析物浓度通过外推法得到.
与偏差有关的不确定度通常主要是外推法的不确定度,以及与(适合时)来自制备和添加贮存溶液的重要的不确定度分量一起合成.
注:为了直接相关,该加入应直接加到最初样品,而不是已制备的萃取物中.
7.
7.
8对于所有已知的和显著的系统影响必须进行校正,这是ISO指南中的通用要求.
当对一个显著的总体偏差进行修正时,与偏差有关的不确定度的评估按照7.
7.
5节所描述的有关处理不显著的偏差的情况进行.
7.
7.
9偏差显著时,但是又因为某个实际的原因忽略不计了,此时必须采取其它的措施(见7.
15节).
其他因素7.
7.
10剩下的其他因素的影响应分别估计.
可以通过实验变化的方法,也可通过已建理论预测的方法进行评估.
与这些因素相关的不确定度也应该加以评估、记录、并和其他分量按照常规方法合成.
7.
7.
11与该研究的精密度相比,这些剩余因素的影响被证明可以忽略不计时(即统计上非显著时),建议其不确定度分量等于与该因素有关的显著性检验的标准偏差.
例:对所允许的一个小时萃取时间变化的影响通过t-检验进行了研究,对于正常萃取时间和减少一个小时的萃取时间分别对同一样品测试了五次.
平均值和标准偏差(用mglP-1P表示)是:标准时间:平均值为1.
8,标准偏差为0.
21;而另一个时间:平均值为1.
7,标准偏差为0.
17.
t-检验使用合并方CNAS-GL06:2006第26页共148页2006年06月01日发布2006年07月01日实施差以获得t值,其中合并方差等于82.
05151037.
0)7.
18.
1(037.
0)15()15(17.
0)15(21.
0)15(22=+*==+*+*t与3.
2=critt相比,这是不显著的.
但注意该差值(0.
1)是与所计算的标准差术语来比较的,其值=12.
0)5/15/1(037.
0=+*.
该值就是与萃取时间允许变化的影响有关的不确定度分量.
7.
7.
12当测到某一个影响在统计学上是显著时,但是在实践中又足够小可以忽略不计时,参考7.
15节的方法处理.
7.
8经验方法的不确定度评估7.
8.
1'经验方法'是指在特定应用领域内,为测量比较的目的而一致同意使用的方法,其特征是被测量依赖于所使用的方法.
该方法因此定义了被测量.
例子包括从陶瓷中溶出金属的测定方法和食物中的食用纤维的测定方法(也见5.
2节和例A5).
7.
8.
2当此类方法用于其指定领域时,其方法偏差规定为零.
在这种情况下,偏差估计中应只和实验室的操作相关,不应额外考虑方法内在的偏差.
含义如下所述.
7.
8.
3无论是为了证明偏差可忽略还是为了测量偏差,进行标准物质研究时应使用经特定方法验证的标准物质或使用其值已该通过特定方法获得以供比较的标准物质.
7.
8.
4当具有此类特征的标准物质不能获得时,对偏差的整体控制就与对影响结果的方法参数控制相关,特别是时间、温度、质量、体积等因素.
与这些输入因素有关的不确定度因此必须进行评估,或证明其可以忽略不计,或对其定量化(见例子A6).
7.
8.
5经验方法通常作为协同研究对象,所以应按照7.
6节中方法评估其不确定度.
7.
9特别方法(ad-hocmethods)的不确定度评估7.
9.
1特别方法是在短期内或为一小批测试样而进行的探测性研究所建立的方法.
这种方法通常基于标准方法或实验室内制定的良好方法,但做了大的修改(例如研究不同的被分析物)并且通常没有合理的理由为所讨论的特定材料进行正式的确认研究.
7.
9.
2由于为建立有关不确定度分量所付出的努力有限,有必要大部分依赖于有关系统已知性能或这些系统内的有关部分的已知性能.
不确定度评估因此就应该基于一个有关系统CNAS-GL06:2006第27页共148页2006年06月01日发布2006年07月01日实施或多个系统的已知性能.
该性能信息必须被必要的研究所支持,以确认信息的相关性.
下面建议假定存在这样一个相关的系统,并且为获得可信的不确定度估计值已进行了充分的研究,或该方法包括了其他方法的部分,并且这些部分的不确定度以前已经确定.
7.
9.
3至少,有必要得到所讨论方法的总偏差的估计值和精密度的表示值.
测量偏差理想的做法是用标准物质,但实际上更常用的做法是通过加料回收率加以评估.
除了加料回收率要与相关系统内所观察的值相比较,以建立以前研究与所讨论的特别方法之间联系外,7.
7.
4节的讨论适用.
对所测试材料,在本研究要求内,特别方法所观察的总偏差,应与有关系统的观察值相当.
7.
9.
4最少的精密度试验由一次重复分析组成.
然而,我们推荐进行尽可能多的重复分析.
该精密度应与相关系统的精密度比较.
特别方法的标准偏差应相当的.
注:建议比较应基于检查.
统计上的显著性检验(如F-检验)对于重复次数小的重复分析通常不可靠,并且仅仅因为该检验的不充分将易于导致'不存在显著差异'的结论.
7.
9.
5当上述条件明确得到满足时,相关系统的不确定度评估值可以直接应用到特别方法所得的结果中,并根据浓度和其他已知因素进行适当调整.
7.
10单个分量的量化7.
10.
1几乎总是很有必要单独考虑一些不确定度的来源.
在有些情况下,只需考虑很少量的来源,在另一些情况下,特别是方法性能数据很少或没有时,应分别研究每个来源(见附录A的例1、2和3的说明).
有几个通用的方法来建立单个的不确定度分量:·输入变量的实验变化·根据现有数据,例如测量和校准证书·通过理论原则建立的模型·根据经验和假设模型的信息作出的判断这些不同的方法将分别简述如下.
7.
11单个不确定度分量的试验估计7.
11.
1通过针对特定参数的试验研究来进行不确定度分量的估计,通常是可能和可行的.
7.
11.
2随机影响的标准不确定度通常通过重复性试验测量,并以被测量值的标准偏差的形式定量表示.
实践中,通常重复试验不超过十五次,除非需要更高的精密度.
7.
11.
3其他典型的试验包括:CNAS-GL06:2006第28页共148页2006年06月01日发布2006年07月01日实施·单个参数变化对结果影响的研究.
尤其适合于独立于其他影响的连续、可控制参数的情况,如时间或温度.
结果随参数变化的变化率可通过实验数据获得.
然后将其直接与该参数的不确定度合成就可得到相关的不确定度分量.
注:与该研究现有精密度相比,参数的变化应该足够使结果产生大的变化.
(例如,重复测量的标准偏差的五倍).
·稳健性研究,系统地研究参数适当变化的显著性.
尤其适合于快速识别显著影响;并且通常用于方法优化.
该方法可用于离散影响的情况,如基体的变化,或小型仪器结构的变化,它们可能对结果产生不可预测的影响.
当发现某个因素是显著的,通常要进一步研究.
当不显著时,与之有关的不确定度是稳健性研究所得不确定度(至少对初步评估是这样).
·系统地多参数实验设计方案旨在评估因素影响和相互作用.
这类研究在研究绝对变量时尤其有用.
绝对变量是其值与影响量的大小无关的参数.
在一个研究中的实验室数目、分析人员名字或样品类型就是绝对变量的例子.
例如,基体类型变化(在所述方法范围内)的影响可从重复的多基体研究中所进行的回收率研究中估计.
然后,方差分析就能提供所观察到的分析回收的基体内和基体间方差分量.
基体间方差分量能够提供与基体变化有关的标准不确定度.
7.
12基于其他结果或数据的评估7.
12.
1通常可能使用任何现有的关于所考虑量的不确定度的相关信息来评估某些标准不确定度.
下面建议一些信息来源.
7.
12.
2U水平测试(PT)项目U.
实验室参加水平测试的结果可以用于检查已评估的不确定度,因为该不确定度应该与该实验室参加多个回合的水平测试的结果的分布兼容.
特别是在下述特殊情况下:·当水平测试项目所用样品的组成覆盖日常分析的全部范围·当每个回合的所赋值可追溯到适当的参考值,以及·所赋值的不确定度比观察到的结果分布小此时,在重复回合中报告值和所赋值之差的分散较好地为评估在水平测试项目范围内测量过程所产生的测量不确定度提供了基础.
例如,对于具有类似材料和被分析物水平的含量测试项目,结果之差的标准偏差给出了标准不确定度.
当然,对于可溯源的所赋值的CNAS-GL06:2006第29页共148页2006年06月01日发布2006年07月01日实施系统偏差和不确定度的其他来源(如7.
6.
1节中所指的那些)也必须加以考虑.
7.
12.
3U质量保证(QA)数据U:如前所述,必须保证达到标准操作程序中所制定的质量准则.
并且对质量保证样品的测量显示该准则继续得到满足.
在质量保证核查中使用标准物质时,7.
5节中给出了如何使用该数据来评估不确定度.
如果使用了任何其他稳定的物质,质量保证数据提供了一个中间精密度的估计值(7.
7.
2节).
质量保证数据也构成了不确定度所引用数值的持续核查.
很显然,由随机影响产生的合成不确定度不可能小于质量保证测量的标准偏差.
7.
12.
4U供应商的信息U:对于许多不确定度来源,校准证书或供应商的目录可以提供信息.
例如:容量玻璃器皿在使用前,其允差可由生产厂商的产品目录上取得,或从针对特定项目的校准证书上取得.
7.
13根据理论原理建立模型7.
13.
1很多情况下,得到公认的物理理论可以提供结果影响量的良好模型.
例如:温度对体积和密度的影响已了解的很清楚.
在这种情况下,可以使用第8节的不确定度传播方法从关系式评估或计算不确定度.
7.
13.
2在其他情况下,可能有必要将近似的理论模型跟实验数据一起使用.
例如,当分析测量取决于需计时的衍生反应时,可能有必要评估与计时有关的不确定度.
可简单地通过变化所用的时间来达到.
然而,可通过对在所测量浓度附近的衍生动力学的简要实验研究来建立近似的速率模型,并且在给定的时间从所预测的变化速率来评估不确定度,这样做更好.
7.
14基于判断的评估7.
14.
1不确定度的评估既不是日常工作,也不是纯数学推导.
它依赖于对被测量本质和所采用的测量方法和程序的详细了解.
对于作为测量结果所引用的不确定度,其质量和使用最终依赖于对其数值赋值有贡献的那些因素的理解、严格分析和完整性.
7.
14.
2很多数据的分布可以理解为较少观察到其在边界的情况,而较多观察到在中间的情况.
对于这些分布及其相关标准偏差的定量可通过重复测量得到.
7.
14.
3然而,不能重复测量或不能提供特定不确定度分量的有意义测量时,可能需要对于区间的其他评估.
7.
14.
4在分析化学中,有很多例子,后者占主要的情况,因此需要用判断来评估.
例如:CNAS-GL06:2006第30页共148页2006年06月01日发布2006年07月01日实施·不能对每个单一样品进行回收率和其不确定度的评估.
相反,只能对样品类型作出评估(例如,按基体的类型分组),并且结果适用于所有相似类型的样品.
类似程度其本身是一个未知量,因此该推论(从某一类型的基体到某一特定样品)是与不确定度的一个额外分量有关,而对其通常没有说明.
·由分析程序中的规定所定义的测量模型是用来将被测数量值转换为被测量的值(分析结果).
该模型,如科学上的所有模型一样,本身也有不确定度.
只是假设其按特定模型运作,但永远不能100%肯定.
·使用标准物质是极其鼓励的,但仍有不确定度,不仅其真值有不确定度,分析具体样品时特定标准物质的相关性也有不确定度.
因此,在特定情况下需要判断所声明使用的标准物质与样品性质合理接近的程度.
·当程序对被测量定义不充分时会产生另一种不确定度来源.
比如测定"可氧化的高锰酸盐物质"的情况,无论是分析地上的水或是城市废水,毫无疑问都是不同的.
不仅有氧化等因素,基体组成或干扰等化学影响均可能对该技术规定有影响.
·在分析化学中通常的做法是加入单一物质,如结构接近的类似物或同位素标记化合物,相关的内在物质的回收率或甚至整类的化合物的回收率可据此判断.
很明显,假如分析家准备研究在所有浓度水平,以及任何比例的被测量对加入物和所有"相关"基体的回收率,则其不确定度可通过实验评估.
但通常避免进行该实验,而采用下列判断:*浓度与被测量回收率的相关性*浓度与加入物回收率的相关性*回收率与基体(次)类型的相关性*内在物质和加入物的结合模型的性质7.
14.
5此类判断不是基于直接的试验结果,而是基于主观(人为的)的概率,这是一个与'可信程度'、'直觉概率'和'可信性'[H.
11]同义的术语.
也假定可信程度不是基于仓促的判定,而是周密考虑并成熟的概率判定.
7.
14.
6虽然主观概率因人而异,甚至对于同一个人因时而异,这是公认的,但并不武断,因为他们受常识、专业知识和以往经验和观察的影响.
7.
14.
7这可能是一个短处,但是在实际中不必导致比重复测量更坏的评估.
当实验条件的真实、实际的变化性不可模拟、以及因此导致的结果数据的变化不能反映实际情况时特别适用.
CNAS-GL06:2006第31页共148页2006年06月01日发布2006年07月01日实施7.
14.
8当需要对长期的变化性进行评估,而又没有协同研究数据时,就会产生这种性质的典型问题.
科学家没有选择用主观概率代替实际被测概率(当后者不能得到时),就可能忽略了合成不确定度的重要分量,也就最终不如那些利用主观概率的人客观了.
7.
14.
9评估合成不确定度时,可信程度的两个特征是最基本的:·可信程度应被视为区间值,即提供了类似于经典概率分布的上下边界,·将不确定度的'可信程度'分量合成为合成不确定度,同其它方法得到的标准偏差一样,用同样的计算规则.
7.
15偏差的显著性7.
15.
1对所有的已知并显著的系统影响进行修正,这是ISO指南的总要求;7.
15.
2在判定一个已知的偏差是否可合理忽略不计时,建议使用下列方法:ⅰ)在不考虑相关偏差的情况下评估合成不确定度;ⅱ)将偏差与合成不确定度比较;ⅲ)当偏差与合成不确定度比较不显著时,该偏差可以忽略不计;ⅳ)当偏差与合成不确定度比较显著时,需要采取额外的措施.
合适的措施可能包括:·消除或修正偏差,并适当考虑该修正的不确定度.
·除结果外,还要报告所观察到的偏差及其不确定度注:当已知偏差未能按惯例修正时,该方法应视为经验方法(见7.
8节).
8.
第四步计算合成不确定度8.
1标准不确定度8.
1.
1合成前,所有不确定度分量必须以标准不确定度即标准偏差表示.
这可能涉及到将某些度量分散性的其他方法进行转换.
下面的规则为将一个不确定度分量转换成为标准不确定度提供了某些指导原则.
8.
1.
2当不确定度分量是通过试验方法用重复测量的分散性得出时,可以容易用标准偏差的形式表示.
对于单次测量的不确定度分量,标准不确定度就是所观察的标准偏差;对于平均值的结果,使用平均值的标准差【B.
24】.
8.
1.
3当不确定度的评估是源于以前的结果和数据时,可能已经用标准偏差的形式表示了.
如果给出了带有置信水平的置信区间(用±α表示,并指明p%),则将α值除以与所给出CNAS-GL06:2006第32页共148页2006年06月01日发布2006年07月01日实施的置信水平相应的正态分布百分点的值就得到标准偏差.
例子:规范中规定天平的读数为±0.
2mg,置信水平为95%.
从正态分布的百分点标准表上,95%的置信区间用1.
96σ值进行计算.
使用这个数值得出标准不确定度约为(0.
2/1.
96)≈0.
1.
8.
1.
4如果限值±α给出时没有给定置信水平,有理由认为可能是极限值,通常假定其为矩形分布,标准偏差为3/α(见附录E).
例子:证书给出10mlA级容量瓶为±0.
2ml,则该标准不确定度为ml12.
03/2.
0≈.
8.
1.
5如果±α的限值给出时没有给定置信水平,但是有理由认为不可能是极限值,通常假定为三角形分布,标准偏差为6/α(见附录E).
例子:证书给出10mlA级容量瓶为±0.
2ml,但日常内部检查表明极限值的可能性极少.
则标准不确定度为ml08.
06/2.
0≈.
8.
1.
6当基于判断作出评估时,可能将分量直接评估为标准偏差.
如果不可能,应对现实可能合理存在的最大偏差(剔除简单错误)进行评估.
如果较小值更加可能时,这一估计应按三角形分布处理.
如果没有理由相信小误差比大误差更加可能时,应按矩形分布处理.
8.
1.
7最常用的分布函数的转换因子见附录E.
1.
8.
2合成标准不确定度8.
2.
1在评估了单个的或成组的不确定度分量并将其表示为标准不确定度后,下一步就是要使用下列程序之一计算合成标准不确定度.
8.
2.
2数值y的合成标准不确定度)(yuc和其所依赖的独立参数nxxxK,,21的不确定度之间的总关系式如下:∑∑====niiniiicxyuxucxxyu,12,12221),()(.
.
.
)),((**ISO指南应用更简短的形式)(yui代替),(ixyu.
其中),,(21Kxxy为几个参数K,,21xx的函数,cBiB为灵敏系数,记做iixyc=/,y对xCNAS-GL06:2006第33页共148页2006年06月01日发布2006年07月01日实施的偏导.
),(ixyu为由ix不确定度引起的y的不确定度.
每个变量的分量),(ixyu正好是其用标准偏差表达不确定度的平方乘以相关的灵敏系数的平方.
这些灵敏系数反应了y值如何随着参数21,xx等的变化而变化.
注:灵敏系数也可以直接根据实验得出,当无法找到可靠的数学表达式时极为有用.
8.
2.
3变量相关时,关系式更复杂了:∑∑≠==+=kinlkikikiniiijixxuccxucxyu,,,122.
.
.
,),()())((其中),(kixxu是ix和kx之间的协方差,ic和kc是8.
2.
2节所描述和评估的灵敏系数.
协方差与相关系数iKr有关11,)()(),(≤≤=ikikkikirrxuxuxxu其中8.
2.
4无论不确定度是与单个参数有关,还是与组参数有关,或者是与整个方法有关,上述通用程序都适用.
然而,如果不确定度分量与整个程序有关时,通常表示为对最终结果的影响.
在这种情况下,或当某个参数的不确定度直接表示为其对y的影响时,灵敏系数等于1.
0.
例子:结果为122mgl,其标准偏差为11.
4mgl.
在这些条件下,与精密度有关的标准不确定度u(y)为11.
4mgl.
为清楚起见忽略其他因素,则该测量所蕴含的模型为y=(计算的结果)+ε其中ε代表了测量条件下的随机变化影响.
因此ε/y等于1.
08.
2.
5除了上述情形,当灵敏系数等于1外,以及除了下列规则1和规则2中所给的特殊例子外,必须使用需要进行偏导或数值当量的本通用程序.
附录E中给出了由Kragten提出的数字计算方法的细节[H.
12],该方法有效利用了电子表格软件,根据已知测量模型,从输入标准不确定度得出合成标准不确定度.
除最简单的情况外,建议使用本方法和其他适合的计算机方法.
8.
2.
6有时,合成不确定度的表达可以采用更为简单的形式.
合成标准不确定度的两个简单规则如下:CNAS-GL06:2006第34页共148页2006年06月01日发布2006年07月01日实施U规则1U:对于只涉及量的和或差的模型,例如:y=(p+q+r+.
.
.
),合成标准不确定度)(yuc如下:.
.
.
.
.
.
)()(.
.
.
)),((22++=qupuqpyucU规则2U:对于只涉及积或商的模型,例如y=(p*q*r*…)或y=p/(q*r*…),合成标准不确定度)(yuc如下:.
.
.
.
.
.
)()()(22++=qquppuyyuc其中,u(p)/p等是参数表示为相对标准偏差的不确定度.
注:减法的处理原则与加法相同,除法与乘法相同.
8.
2.
7合成不确定度分量时,为方便起见,应将原始的数学模型分解,将其变为只包括上述原则之一所覆盖的形式.
例如:表达式(o+p)/(q+r)应分解成两个部分:(o+p)和(q+r).
每个部分的临时不确定度用规则1计算,然后将这些临时不确定度用规则2合成为合成标准不确定度.
8.
2.
8下述例子说明了上述规则的使用.
例1:y=(p-q+r),其中p=5.
02,q=6.
45,r=9.
04,标准不确定度u(p)=0.
13,u(q)=0.
05,u(r)=0.
22.
则,y=5.
02-6.
45+9.
04=7.
6126.
022.
005.
013.
0)(222=++=yu例2:y=(op/qr),其中o=2.
46,p=4.
32,q=6.
38,r=2.
99,标准不确定度u(o)=0.
02,u(p)=0.
13,u(q)=0.
11,u(r)=0.
07.
则y=(2.
46*4.
32)/(6.
38*2.
99)=0.
56024.
0043.
056.
0)(99.
207.
038.
611.
032.
413.
046.
202.
056.
0)(2222=*=+++*=yuyu8.
2.
9很多情况下,不确定度分量的大小随着分析物水平的不同而变化.
例如,回收率的CNAS-GL06:2006第35页共148页2006年06月01日发布2006年07月01日实施不确定度对于高浓度的物质可能要小些,光谱信号可能会近似按与密度相称的比例随机地变化(恒定变化系数).
在这种情况下,必须考虑合成不确定度随被分析物的浓度变化.
方法包括:z将规定的程序或不确定度评估限制到被分析物浓度的小范围内.
z将不确定估计值以相对标准偏差的形式给出.
z明确计算相关性并对给定的结果重新计算不确定度;附录E4中给出了这些方法的其他信息.
8.
3扩展不确定度8.
3.
1最后一步是将合成标准不确定度和所选的包含因子相乘得到扩展不确定度.
扩展不确定度需要给出一个期望区间,合理地赋予被测量的数值分布的大部分会落在此区间内.
8.
3.
2在选择包含因子k的数值时,需要考虑很多问题,包括:·所需的置信水平;·对基本分布的了解;·对于评估随机影响所用的数值数量的了解(见下面的8.
3.
3节).
8.
3.
3大多数情况下,推荐k为2.
然而,如果合成不确定度是基于较小自由度(大约小于6)的统计观察的话,选择这个k值可能不充分.
此时k的选择取决于有效自由度.
8.
3.
4当合成标准不确定度由某个自由度小于6的分量占决定作用时,推荐将k设成与该分量自由度数值以及置信水平(通常95%)相当的学生t分布的双边数值.
表1中给出了t的主要数值.
例子:称量操作的合成标准不确定度,由标准不确定度分量mgucal01.
0=和5次重复实验的标准偏差mgsobs08.
0=合成.
合成标准不确定度mguc081.
008.
001.
022=+=.
很明显基于5次重复性实验并具有5-1=4自由度的重复性分量sBobsB占主要.
因此k要基于学生t分布.
对于自由度为4,置信水平为95%,查表双边数值t为2.
8;因此k值设为2.
8,扩展不确定度U=2.
8*0.
081=0.
23mg.
8.
3.
5对于使用少量的测量来评估大的随机影响,ISO指南[H.
2]对选择k值的选择方法给出了额外的指导,当评估有几个显著分量的自由度时应参照此指南来执行.
8.
3.
6当所涉及的分布是正态分布时,包含因子2(或根据第8.
3.
3-8.
3.
5节选择的包含因子,置信水平设为95%)给出了大约包含95%分布数值的区间.
在没给出分布的情况下,该区CNAS-GL06:2006第36页共148页2006年06月01日发布2006年07月01日实施间并不意味着是在95%的置信水平下的区间.
表1:95%置信(双边)的学生t分布自由度νt112.
724.
333.
242.
852.
662.
59.
不确定度的报告9.
1总则9.
1.
1报告测量结果所需要的信息取决于其预期的用途.
本指南原则如下:·提供足够的信息以便有新的信息或数据时重新评价结果;·多提供些信息总比少提供信息要好.
9.
1.
2当测量的详细情况,包括如何确定不确定度,主要来自已出版的文件时,必须保证所使用的文件是最新的,并且与所使用的方法一致.
9.
2所需要的信息9.
2.
1测量结果的完整报告应包括,或者引用包括下列信息的文件:·根据实验观察值及输入数据进行测量结果及其不确定度计算的方法描述.
·在计算和不确定度分析中使用的所有修正值和常数的数值和来源.
·所有不确定度分量的清单,包括每一个分量是如何评价的完整文件.
9.
2.
2数据和分析的表达方式应能在必要时容易地重复所有重要步骤并重复计算结果:9.
2.
3当需要详尽的报告,包括中间输入数值时,报告应:·给出每一个输入值的数值及其标准不确定度和获取方法的描述.
·给出结果和输入值之间的关系式及其任何偏导数、协方差或用来说明相关性影响的相关系数;·给出每个输入值的标准不确定度的自由度的评估值.
(评估自由度的方法见ISO指CNAS-GL06:2006第37页共148页2006年06月01日发布2006年07月01日实施南【H.
2】).
注:当函数关系极其复杂或没有明确的函数关系时(例如,可能仅以计算机程序的形式存在),可以用通用术语或引用适当的文献来描述上述关系.
在这种情况下,必须清楚地说明结果值及其不确定度是如何得到的.
9.
2.
4报告日常分析的结果时,仅给出扩展不确定度的数值和k值就可能足够了.
9.
3报告标准不确定度9.
3.
1当不确定度是以标准不确定度uBcB的形式表达时(即一个标准偏差),推荐采用下面的形式:"(结果):x(单位)[和]标准不确定度uBcB(单位)(其中该标准不确定度是国际计量学基本和通用术语词汇表(第二版,ISO1993年)所定义的标准不确定度,相当于一个标准偏差)注:当使用标准不确定度时,不建议使用±符号,因为该符号通常与高置信水平的区间有关.
括号[]中的术语根据需要可以忽略或精简.
例子:总氮含量:3.
52%w/w标准不确定度:0.
07%w/w**标准不确定度相当于一个标准偏差9.
4报告扩展不确定度9.
4.
1除非另有要求,结果x应跟使用包含因子k=2(或按8.
3.
3节所描述,给出k值)计算的扩展不确定度U一起给出.
推荐采用以下方式:"(结果):(x±U)(单位)[其中]报告的不确定度[国际计量学基本和通用术语词汇表(第二版ISO1993年)所定义的扩展不确定度]计算时使用的包含因子为2,[其给出了大约95%的置信水平]"括号[]中的术语可以适当省略或精简.
当然,包含因子应加以调整以反映实际使用的数值.
例子:总氮量:(3.
52±0.
14)%w/w**报告的不确定度是扩展不确定度,使用的包含因子是2,对应的置信水平大约是95%.
CNAS-GL06:2006第38页共148页2006年06月01日发布2006年07月01日实施9.
5结果的数值表示9.
5.
1结果及其不确定度的数值表示中不可给出过多的数字位数.
无论是给出扩展不确定度U,还是标准不确定度u,通常不确定度的有效数字不要多于两位的.
测试结果应根据所给出的不确定度进行适当修约.
9.
6与限值的符合性9.
6.
1被测量(例如:有毒物质的含量)法规通常要求被证明在规定限值内.
本文中的测量不确定度明显地有解释分析结果的含义,特别是:·评价符合性时可能需要考虑分析结果的不确定度;·限值中可能已经给出不确定度的允许量.
在评估中需考虑这两个因素.
下面给出了常规的做法.
9.
6.
2假定限值设定时没有给出不确定度的允许值,对于与上限的符合性情况明显有四种情形(见图2):i)结果高出限值超过一个扩展不确定度;ii)结果高出限值,但不超过一个扩展不确定度;iii)测量结果低于限值,但不超过一个扩展不确定度;iv)结果低于限值超过一个扩展不确定度情形i)通常解释为完全不符合.
情形iv)通常解释为符合.
情形ii)和iii)通常需要根据与数据用户的协议分别考虑.
对于与低限符合性的情况,类似的论点适用.
9.
6.
3当已知或相信设定限值时已考虑到不确定度的允许量时,符合性判断有理由只对给出的允许量进行.
当符合性检查是对照在指定的环境中操作的规定方法时,将产生例外的情况.
在该要求中蕴含着一个假设,即该规定方法的不确定度,或至少是复现性,是足够小以至在实际中可以忽略.
在这种情况下,如果提供了适当的质量控制,通常只对特定结果的数值报告符合性.
这通常会在采用本方法的标准中声明.
CNAS-GL06:2006第39页共148页2006年06月01日发布2006年07月01日实施上控限(i)(ii)(iii)(iv)结果高于限值超过结果高于限值但不结果低于限值但不结果低于限值超一个不确定度超过一个不确定度超过一个不确定度过一个不确定度图2不确定度和符合性限值CNAS-GL06:2006第40页共148页2006年06月01日发布2006年07月01日实施附录A例子介绍简介这些例子阐明了第5至7节所描述的不确定度评估技术如何应用到某些典型的化学分析中.
它们均按照流程图所给出的步骤进行.
识别不确定度来源并在因果图中列出(见附录D).
这有助于避免不确定度来源的双重计算,也有助于将能评估合成效应的分量组合在一起.
例1-6阐明了应用附录E.
2的电子表格方法从已计算出的分量),(ixyu计算合成不确定度.
P*P第8.
2.
2节解释了分量u(y,xBiB)计算的原理.
例1-6的每一个例子均有一个介绍性的摘要.
给出了分析方法的概要、不确定来源表格及其各自的分量、不同分量的图形比较、以及合成不确定度.
例1-3分别阐明了通过量化每一个来源的不确定度来估算不确定度.
每一个均给出了与使用容量玻璃器皿进行体积测量和不同称重方法称量质量有关的不确定度的详细分析.
该细节是为了说明目的,不应作为所需详细程度或所采用的方法的通用建议.
对于许多分析,与这些操作有关的不确定度不是显著的,这样详细的评估将是没必要的.
使用这些操作的典型值并适当考虑所使用的质量和体积的实际值就足够了.
例子A1例子A1是一个为AAS制备HNOB3B中镉的标准溶液的非常简单例子.
其目的是说明如何评估体积测量和称重的基本操作所引入的不确定度分量,以及这些分量如何合成得出总的不确定度.
例子A2例子A2是一个通过标准邻苯二甲酸氢钾(KHP)滴定测量来制备氢氧化钠(NaOH)标准溶液的例子.
它包括了例A1所述的简单体积测量和称量的不确定度评估和对与滴定测量有关的不确定度检查.
例子A3例子A3是例A2的延伸,其增加了用制备好的NaOH滴定HCl.
例子A4CNAS-GL06:2006第41页共148页2006年06月01日发布2006年07月01日实施例子A3说明了按7.
7节所述使用内部确认数据,以及如何使用这些数据来评估许多来源的合成效应所引起的不确定度.
也说明了如何评估与方法偏差有关的不确定度.
例A5例子A5说明了按7.
2-7.
8节所述,使用规定程序测量陶瓷上溶出的重金属含量,如何评估这种按标准或"经验"方法结果的不确定度.
其目的是说明如何在缺乏协同实验数据或稳健性试验结果的情况下,有必要考虑方法定义所允许的参数(例如温度、浸泡时间和酸度)范围所引起的不确定度.
当可获取协同研究数据时,该过程变得相当简单,如下一个例子所示.
例A6例子A6是粗(食用)纤维测定的不确定度评估.
因为只使用标准方法所定义的被分析物,所以该方法是经验的方法.
在这个例子中,协同研究数据、内部质量保证核查以及文献研究数据均是现成的,允许使用7.
6节所描述的方法.
内部研究证实该方法的性能达到以协同研究所预期的结果.
该例子说明了使用内部方法性能核查所支持的协同研究数据如何能大大地降低在此情况下不确定度评估所需要的不同分量的数目.
例A7例子A7详细说明了使用IDMS测量水样中铅含量的不确定度评估.
除了识别可能的不确定度来源并用统计方法量化之外,该例子说明了如何有必要按7.
14节所述的基于判断来评估不确定度分量.
使用判断是ISO指南[H.
2]所描述的B类评估的一个特定例子.
CNAS-GL06:2006第42页共148页2006年06月01日发布2006年07月01日实施例子A1:校准标准溶液的制备概要目的由高纯金属(镉)制备浓度约为11000mgl的校准标准溶液.
测量步骤清洁高纯金属的表面以便除以任何金属氧化物的污染.
然后称量金属并将金属溶于容量瓶的硝酸中.
该步骤的各个阶段见下述流程图.
被测量:][.
.
10001=mglVpmCcd其中:cdC:校准标准溶液的浓度][1mgl1000:从[ml]到[l]的换算系数m:高纯金属的质量[mg]p:以质量分数给出的金属纯度V:校准标准溶液的溶液体积不确定度来源的识别有关的不确定度来源见下页的因果图.
不确定度分量的量化数值及其不确定度见表A1.
1.
合成标准不确定度制备17.
1002mgldC校准标准溶液的合成标准不确定度为19.
0mgl.
对合成标准不确定度的不同分量的大小在图A1.
2中以图表形式列出.
清洁金属表面称量金属溶解并稀释结果图A1.
1镉标准溶液的制备CNAS-GL06:2006第43页共148页2006年06月01日发布2006年07月01日实施表A1.
1数值和不确定度描述数值标准不确定度相对标准不确定度u(x)/xp金属纯度0.
99990.
0000580.
000058m金属质量100.
28mg0.
05mg0.
0005V量瓶体积100.
0ml0.
07ml0.
0007cdc校准标准溶液的浓度17.
1002mgl19.
0mgl0.
0009)()/(),(iiixuxyxyu=的数值取自表A1.
3可读性可读性温度校准复现性V纯度c(Cd)m(tare)m(gross)线性复现性复现性线性校准m校准灵敏度灵敏度Vm纯度c(Cd)00.
20.
40.
60.
81u(y,xB1B)(mgГP1P)图A1.
2在制备镉标准溶液的不确定分量CNAS-GL06:2006第44页共148页2006年06月01日发布2006年07月01日实施例A1:校准标准溶液的制备详细讨论A1.
1引言第一个介绍的例子讨论了从相应的高纯金属制备原子吸收光谱(AAS)所使用的校准标准溶液(本例子中在稀3HNO中约为11000mgl).
尽管该例子没有给出完整的分析测量过程,但校准标准溶液的使用几乎是每一次测量的组成部分,因为现代日常分析测量都是比较测量,需使用参考标准以溯源至SI.
A1.
2步骤1:技术规定该第一步骤的目的是写出被测量的清楚描述.
该技术规定包括校准标准溶液制备的描述以及被测量和其所依赖的参数之间的数学关系.
程序关于如何制备校准标准溶液的具体信息通常在标准操作程序(SOP)中给出.
制备由下列各阶段组成:各步骤是:ⅰ)高纯金属的表面用酸混合物来处理以除去任何金属氧化物污染.
清洁方法由金属制造商提供,并且需要照着去做以便获得证书上所声明的纯度.
ⅱ)分别称量容量瓶(100ml)的重量和容量瓶与净化的金属重量.
使用的天平具有0.
01mg的分清洁金属表面称量金属溶解并稀释结果图A1.
3镉标准溶液的制备CNAS-GL06:2006第45页共148页2006年06月01日发布2006年07月01日实施辨率.
ⅲ)1ml的硝酸(65%m/m)和3ml的去离子水加到量瓶中以溶解镉(约100mg,精确称量).
然后用去离子水充满容量瓶直至刻度,并且倒转容量瓶至少30次以充分混合.
计算本例子中的被测量是校准标准溶液的浓度,取决于高纯度金属(Cd)的称重、纯度以及溶解所用的液体体积.
该浓度由下列公式给出:11000=mglVPmccd其中cdC:校准标准溶液的浓度][1mgl1000:从[ml]到[l]的换算系数m:高纯金属的质量[mg]p:以质量分数给出的金属纯度V:校准标准溶液的溶液体积A1.
3步骤2:识别和分析不确定度来源第二步的目的是列出所有影响被测量数值的各个参数的不确定度来源.
U纯度供应商证书上给出的金属(Cd)纯度是99.
99±0.
01%.
因此P是0.
9999±0.
0001.
这些数值取决于清洁高纯度金属表面的有效性.
如果严格按照制造商给出的步骤,就无需将由于金属表面氧化物污染引起的额外不确定度加到证书所给的数值中.
没有信息表明金属100%溶解了.
因此不得不进行重复的制备实验以检查该因素可否被忽略.
U质量m制备的第二阶段涉及到高纯金属的称重.
要制备100ml11000mgl镉溶液.
镉的相应质量由已扣除皮重的称量给出,得出m=0.
10028g.
制造商的说明书确认扣除皮重称量的三个不确定度来源:重复性、可读性(数字分辨率)以及由于天平校准产生的不确定度分量.
该校准操作有两个潜在的不确定度来源,即天平的灵敏度及其线性.
灵敏度可忽略,因为减量称量是用同一架天平在很窄范围内进行.
注:浮力修正不加以考虑,因为所有的称量结果都是按常规在空气中称量[H.
19].
残余的不确定度很小而不加以考虑.
参见附录G的注1.
U体积VCNAS-GL06:2006第46页共148页2006年06月01日发布2006年07月01日实施容量瓶中的溶液体积主要有三个不确定度来源:·确定容量瓶内部体积时的不确定度·充满容量瓶至刻度的变化·容量瓶和溶液温度与容量瓶体积校准时的温度不同不同的因素及其影响见图A1.
4的因果图(见附录D的说明)A1.
4步骤3:不确定度分量的量化在步骤3中,每一个已识别的潜在来源的不确定度的大小,或者使用以前的实验结果直接测量、评估,或者从理论分析导出.
U纯度在证书上给出的Cd的纯度是0.
9999±0.
0001.
由于不确定度数值的其它信息,故假设是矩形分布.
标准不确定度u(P)的值为0.
0001值除以3(见附录E1.
1).
000058.
030001.
0)(==PuU质量m利用校准证书的数据和制造商关于不确定度评估的建议,对Cd质量有关的不确定度进行估算,结果为0.
05mg.
这种评估考虑了前面已识别的三种分量(A1.
3节).
注:质量不确定度的详细计算可能会非常复杂,当质量不确定度是显著的时候,参考可读性可读性温度校准重复性V纯度c(Cd)m(tare)m(mross)线性重复性重复性线性校准m校准图A1.
4Cd标准溶液制备的不确定度灵敏度灵敏度CNAS-GL06:2006第47页共148页2006年06月01日发布2006年07月01日实施制造商提供的资料是很重要的.
在本例中,为简明起见省略了这些计算.
U体积V该体积有三个主要的影响:校准、重复性和温度影响.
ⅰ)校准:制造商提供的容量瓶在20℃的体积为100ml±0.
1ml,给出的不确定度的数值没有置信水平或分布情况信息,因此假设是必要的.
在这里,计算标准不确定度时是假设三角形分布.
mlml04.
061.
0=注:选择三角形分布是因为在一个有效的生产过程中标定值比极限值可能性更高.
最终分布用三角形分布比矩形分布表示更好.
ⅱ)重复性:由于充满容量瓶的变化引起的不确定度可通过该容量瓶的典型样品的重复性实验来评估.
对典型的100ml容量瓶充满10次并称量的实验,得出标准偏差为0.
02ml.
这可直接用作标准不确定度.
ⅲ)温度:根据制造商提供的信息,该容量瓶已在20℃校准,而实验室的温度在±4℃之间变动.
该影响引起的不确定度可通过估算该温度范围和体积膨胀系数来进行计算.
液体的体积膨胀明显大于容量瓶的体积膨胀,因此只需考虑前者即可.
水的体积膨胀系数为14101.
2°*C,因此产生的体积变化为ml084.
0)101.
24100(4±=***±计算标准不确定度时假设温度变化是矩形分布,即mlml05.
03084.
0=三种分量合成得到体积的标准不确定度u(V)mlVu07.
005.
002.
004.
0)(222=++=A1.
5步骤4:计算合成标准不确定度cdc等于][10001=mglVPmccd中间值及其标准不确定度和相对标准不确定度汇总在表A1.
2.
内容数值)(xuxxu)(金属纯度(P)0.
99990.
0000580.
000058金属质量(m)100.
280.
05mg0.
0005量瓶体积V(ml)100.
00.
07ml0.
0007图A1.
2数值和不确定度CNAS-GL06:2006第48页共148页2006年06月01日发布2006年07月01日实施使用这些数值,得校准标准溶液的浓度为17.
10020.
1009999.
028.
1001000=**=mglccd对于这个简单的乘法表达式,与每一个分量有关的不确定度合成如下:112222229.
00009.
07.
10020009.
0)(0009.
00007.
00005.
0000058.
0)()()()(=*=*==++=++=mglmglccuVVummuPPuccucdcdccdcdc使用附录E给出的电子表格方法导出合成标准不确定度))((cdccu更好,因为即使复杂的表达式也能用它.
已填好的电子表格见表A1.
3.
不同参数的分量大小见图A1.
5.
容量瓶体积的不确定度分量是最大的,称量步骤的不确定度分量类似.
镉纯度的不确定度实际上对总的不确定度没有影响.
将合成标准不确定度乘以包含因子2得到扩展不确定度)(cdcU.
118.
19.
02)(=*=mglmglcUcd)()/(),(iiixuxyxyu=的数值取自表A1.
3Vm纯度c(Cd)00.
20.
40.
60.
81u(y,xB1B)(mgГP1P)图A1.
5在制备镉标准溶液的不确定分量CNAS-GL06:2006第49页共148页2006年06月01日发布2006年07月01日实施表A1.
3不确定度的电子表格计算ABCDE1pmV2数值0.
9999100.
28100.
003不确定度0.
0000580.
050.
0745p0.
99990.
9999580.
99990.
99996m100.
28100.
28100.
33100.
287V100.
0100.
00100.
00100.
0789c(Cd)1002.
699721002.
757881003.
199661001.
9983210),(ixyu0.
058160.
49995-0.
701401122),(,)(ixyuyu0.
745290.
003380.
249950.
491961213u(c(Cd)0.
9将参数的值输入到第二行的C2至E2.
其标准不确定度在下面一行(C3-E3).
电子表格将C2至E2值复制到第二列的B5到B7.
使用这些值后得出的结果(c(Cd))见B9.
C5为C2的p值加上其在C3的不确定度.
使用C5-C7数值得出的计算结果见C9.
D和E列按照类似的步骤.
第10行(C10-E10)的值是行(C9-E9)减去B9之值的差.
第10行(C10-E10)值的平方放在第11行(C11-E11),其平方和的结果值在B11.
B13给出合成标准不确定度,也就是B11的平方根.
CNAS-GL06:2006第50页共148页2006年06月01日发布2006年07月01日实施例A2:氢氧化钠溶液的标定概要目的用标准邻苯二甲酸氢钾(KHP)标定氢氧化钠(NaOH)溶液.
测量步骤干燥并称取滴定标准物KHP.
配制NaOH溶液后,将滴定标准物(KHP)溶解,并用NaOH溶液滴定.
具体的测定步骤见流程图A2.
1被测量TKHPKHPKHPNaOHVMPmc=1000[mol/l]其中,NaOHc:NaOH溶液的浓度[mol/l]1000:由[ml]转化为[l]的换算系数HKPm:滴定标准物KHP的质量[g]KHPP:滴定标准物的纯度,以质量分数表示KHPM:KHP的摩尔质量[g/mol]TV:NaOH溶液的滴定体积[ml]不确定来源的识别:图A2.
2的因果关系图标明了不确定度的有关来源.
不确定度分量的量化:表A2.
1列出了各不确定度分量,图A2.
3以图示方式列出.
0.
10214mol/lNaOH溶液的合成标准不确定度为0.
00010mol/l.
称取KHP制备NaOH滴定结果图A2.
1标定NaOHCNAS-GL06:2006第51页共148页2006年06月01日发布2006年07月01日实施表A2.
1:NaOH标定中的数据和不确定度名称数值x标准不确定度u相对标准不确定度u(x)/xRep复现性1.
00.
00050.
0005KHPmKHP的质量0.
3888g0.
00013g0.
00033KHPPKHP的纯度1.
00.
000290.
00029KHPMKHP的摩尔质量24.
2212g/mol0.
0038g/mol0.
000019TVKHP滴定耗用NaOH的体积18.
64ml0.
013ml0.
0007NaOHcNaOH溶液浓度0.
10214mol/l0.
00010mol/l0.
00097终点温度校准复现性V(T)P(KHP)c(NaOH)m(tare)m(mross)线性m(KHP)线性校准M(KHP)校准图A2.
2建立因果图m(KHP)V(T)终点偏差灵敏度灵敏度V(T)00.
02u(y,xB1B)(mmolГP1P)图A2.
3滴定不确定度分量大小M(KHP)P(KHP)m(KHP)重复性c(NaOH)0.
040.
060.
080.
10.
12CNAS-GL06:2006第52页共148页2006年06月01日发布2006年07月01日实施例A2:氢氧化钠溶液的标定详细讨论A2.
1介绍第二个例子讨论的是氢氧化钠(NaOH)溶液浓度的标定实验.
NaOH由滴定标准物邻苯二甲酸氢钾(KHP)标定.
已知NaOH溶液浓度在0.
1mol/l左右.
滴定的终点由自动滴定装置判断,在该装置中装有测定pH曲线形状的组合pH电极.
滴定标准物KHP的有效成分,即与其分子总数有关的自由质子的数目,使标定的NaOH溶液的浓度可溯源至SI国际单位制.
A2.
2步骤1:技术规定步骤1描述了测量的具体步骤,包括列出测定步骤、被测量的数学计算公式及其所依据的参数.
步骤:NaOH溶液的标定包括以下步骤:各步骤是:ⅰ)按供应商的说明,将标准物KHP干燥.
供应商的说明印制在其提供的产品目录中,并且注明了滴定标准物的纯度及其不确定度.
滴定19ml浓度为0.
1mol/l的NaOH溶液大约需要消耗KHP的量为:g388.
00.
11000191.
02212.
204=***称量应使用最后一位为0.
1mg的天平.
ⅱ)配制0.
1mol/l的NaOH溶液.
为配制1升NaOH溶液,大约需要称取4gNaOH.
然而,由于NaOH溶液的浓度是由标准物KHP来测定,而不是直接计算得到,因此不需要与NaOH的分子量或其质量有关的不确定度来源信息.
ⅲ)将称取的滴定标准物KHP溶解于约50ml去离子水中,再以NaOH溶液滴定.
采用自动滴定装置来滴加NaOH溶液,并记录pH曲线.
从自动滴定装置记录的pH曲线判定滴定终点.
计算:被测量,即NaOH溶液的浓度,取决于KHP的质量、纯度、分子量和滴定终点时消耗的NaOH的KHP称重配制NaOH滴定结果图A2.
4氢氧化钠溶液的标定CNAS-GL06:2006第53页共148页2006年06月01日发布2006年07月01日实施体积TKHPKHPKHPNaOHVMPmc=1000[mol/l]其中,NaOHc:NaOH溶液的浓度[mol/l]1000:由[ml]转化为[l]的换算系数KHPm:滴定标准物KHP的质量[g]KHPP:滴定标准物KHP的纯度,以质量分数表示KHPM:KHP的摩尔质量[g/mol]TV:NaOH溶液滴定消耗的体积[ml]A2.
3步骤2:不确定度来源的确定和分析本步骤的目的是确定各主要不确定度来源,了解其对被测量及其不确定度的影响.
本步骤是分析测定的不确定度评估中最困难的,因为一方面有些不确定度来源可能被忽略,另一方面有些不确定度来源可能会被重复计算.
绘制因果图(附录D)是防止这类问题发生的一个可行的方法.
制作因果图的第一步就是先画出被测量计算公式中的四个参数.
然后,分析测定方法的每一步骤,再沿主要影响因素将其它进一步的影响量添加在图中.
对每一个分支干均进行同样的分析,直到影响因素变得微不足道为止,将所有不可忽略的影响均标注在每一个支干上.
U质量UmBKHPBV(T)P(KHP)c(NaOH)M(KHP)图A2.
5建立因果图的第一步m(KHP)CNAS-GL06:2006第54页共148页2006年06月01日发布2006年07月01日实施大约称取388mgKHP来标定NaOH溶液.
称重为减量称量.
因此在因果图上应画出净重称量(mBtareB)和总重称量(mBgrossB)两条支干.
每一次称重都会有随机变化和天平校准带来的不确定度.
天平校准本身有两个可能的不确定度来源:灵敏度和校准函数的线性.
如果称量是用同一台天平且称量范围很小,则灵敏度带来的不确定度可忽略不计.
所有不确定度来源均标注在因果图上(见图A2.
6).
U纯度PUBUKHPUB供应商目录中标注的KHP的纯度介于99.
95%至100.
05%之间.
因此PBKHPB等于0005.
00000.
1±.
如果干燥过程完全按供应商的规定进行,则无其它不确定度来源.
U摩尔质量MUBUKHPUB邻苯二甲酸氢钾(KHP)的传统分子式为CB8BHB5BOB4BK.
该分子的摩尔质量的不确定度可以通过合成各组成元素原子量的不确定度得到.
IUPAC每两年在《纯粹和应用化学杂志》上发表一次包括不确定度评估值的原子量表.
摩尔质量可以直接由该表计算得到;为求简洁,因果图(图A2.
7)省略了各个原子的质量.
U体积VUBUTUB滴定过程借助于20ml的活塞滴定管.
正如前面例子中充满容量瓶一样,NaOH溶液从活塞滴定管滴定的体积有三个同样的不确定度来源.
这三个来源是滴定体积的重复性、体积校准时的不确定度、以及由实验室温度与活塞滴定管校准时温度不一致而带来的不确定度.
此外,终点检测过程也有影响,有两个不确定来源.
1.
终点检测的重复性,它独立于滴定体积的重复性.
重复性V(T)P(KHP)c(NaOH)M(KHP)图A2.
6增加称量步骤不确定度来源的因果图m(KHP)校准校准线性重复性线性m(tare)m(gross)灵敏度灵敏度CNAS-GL06:2006第55页共148页2006年06月01日发布2006年07月01日实施2.
由于滴定过程中吸入二氧化碳及由滴定曲线计算终点的不准确,滴定终点与等当点之间可能存在的系统误差.
以上各项均标明在图A2.
7因果图中.
A2.
4步骤3:不确定度分量的定量步骤2确定的各不确定度来源在步骤3中进行量化,并转化为标准不确定度.
通常,各类实验都至少包含了活塞滴管滴定体积的重复性和称量操作的重复性.
因此,将各重复性分量合并为总试验的一个分量,并且利用方法确认的数值将其量化是合理的,由此导致对因果图的修订,见图A2.
8.
方法确认表明滴定实验的重复性为0.
05%.
该值可直接用于合成不确定度的计算.
U质量UmBKHPBV(T)P(KHP)c(NaOH)M(KHP)图A2.
8因果图(将重复性合并)m(KHP)校准校准线性线性m(tare)m(gross)校准湿度偏差终点重复性V(T)终点m(KHP)灵敏度灵敏度图A2.
7因果图(所有来源)灵敏度灵敏度重复性V(T)P(KHP)c(NaOH)M(KHP)m(KHP)校准校准线性重复性线性m(tare)m(gross)重复性校准温度重复性偏差CNAS-GL06:2006第56页共148页2006年06月01日发布2006年07月01日实施相关称量有:容器和KHP:60.
5450g(观测)容器和减量的KHP:60.
1562g(观测)KHP:0.
3888g(计算)由于引入了前面已经确定的合成重复性,因此没有必要考虑称量的重复性.
天平量程范围内的系统偏移将被抵消.
因此,不确定度仅限于天平的线性不确定度.
线性:天平计量证书标明其线性为15.
0±mg.
该数值是托盘上的实际重量与天平读数的最大差值.
天平制造商自身的不确定度评价建议采用矩形分布将线性分量转化为标准不确定度.
因此,天平的线性分量为mgmg09.
0315.
0=上述分量必须计算二次,一次作为空盘,另一次为毛重,因为每一次称重均为独立的观测结果,两者的线性影响间是不相关的.
由此得到质量mBKHPB的标准不确定度u(mBKHPB)数值为:mgmuKHP13.
0)09.
0(2)(2=*=注1:由于称量均是按常规在空气中进行的,因此不考虑浮力修正[H.
19].
其它不确定度分量太小,不予考虑.
见附录G的注1.
注2:称量滴定标准物时还存在其它问题.
当标准物的温度与天平的温度即使只差1℃,也会产生与重复性分量数量级相当的漂移.
滴定标准物已完全干燥,但称量的环境湿度在约50%的相对湿度时,也会吸收一些湿气.
U纯度UPBKHPBKHPP为1.
0000±0.
0005.
供应商在目录中没有给出不确定度的进一步的信息,因此可将该不确定度视为矩形分布,标准不确定度00029.
03/0005.
0)(==KHPPu.
U摩尔质量UMBKHPB从IUPAC最新版的原子量表中查得的KHP(CB8BHB5BOB4BK)中各元素的原子量和不确定度:元素原子量不确定度标准不确定度C12.
01070008.
0±0.
00046H1.
00794±0.
000070.
000040O15.
9994±0.
00030.
00017CNAS-GL06:2006第57页共148页2006年06月01日发布2006年07月01日实施K39.
0983±0.
00010.
000058对于每一个元素来说,标准不确定度是将IUPAC所列不确定度作为矩形分布的极差计算得到的.
因此相应的标准不确定度等于查得数值除以3.
各元素对摩尔质量的贡献及其不确定度分量为:计算式结果标准不确定度8C0107.
128*96.
08560.
00375H00794.
15*5.
03970.
000204O9994.
154*63.
99760.
00068K0983.
391*39.
09830.
000058上表各数值的不确定度是由前表各元素的标准不确定度数值乘以原子数计算得到的.
KHP的摩尔质量为:molgMKHP/2212.
2040986.
399976.
630397.
50856.
96=+++=上式为各独立数值之和,因此标准不确定度)(KHPMu就等于各不确定度分量平方和的平方根:molgMuKHP/0038.
0000058.
000068.
00002.
00037.
0)(2222=+++=注:由于每个元素对MBUKHPUB的贡献仅是其原子量与原子个数的乘积,那么按照不确定度分量合成的通用规则可以预见每个元素的不确定度可从单个原子分量平方之和来计算,也就是,如对于碳元素,即001.
000037.
08)(2=*=cMu.
然而,请记住该规则只适用于独立的分量,也就是不同测定值的分量.
对于本例,总量是通过单一值乘8得到的.
注意各元素的不确定度分量是独立的,因此可以用常规方式合成.
U体积VUBUTUB1.
滴定体积的重复性:如前所述,该重复性已通过实验合成重复性考虑了.
2.
校准:制造商已给定了滴定体积的准确性范围为±(数值).
对于20ml活塞滴定管,典型数值为ml03.
0±.
假定为三角形分布,标准不确定度为ml012.
06/03.
0=.
注:如果有理由认为出现在中心区的机率大于极值附近时,ISO指南(F.
2.
3.
3)建议采用三角形分布.
例子A1和A2中的玻璃容器均假定为三角形分布(见例子A1中体积不确定度的讨论).
3.
温度:由于对温度缺乏控制而产生的不确定度按前例方式计算,但这一次假定温度的波动范围CNAS-GL06:2006第58页共148页2006年06月01日发布2006年07月01日实施为3±℃(置信水平为95%).
同样用水的膨胀系数4101.
2*℃1得到ml006.
096.
13101.
2194=***因此因温度控制不充分而产生的标准不确定度为0.
006ml.
注:当处理由于环境因素,如温度控制不充分而产生的不确定度时,考虑不同中间值的影响因素之间的相关性是很重要的.
本例中,溶液温度的主要影响是考虑不同溶剂不同的热效应,也就是说溶液与周围温度并不平衡.
因此,本例中,在STP(标准温度和压力)下温度对每种溶液浓度的影响是不相关的,因此作为独立不确定度分量处理.
4.
终点检测误差:滴定是在氩气层下进行的以避免滴定液吸收COB2B带来的误差.
这样的做法就是尽量避免任何偏差,以免修正.
由于是强碱滴定强酸,没有迹象表明由从pH曲线形状判定终点会与等当点不一致.
所以终点判定偏差及其不确定度可以忽略.
VBTB求得为18.
64ml,合并各不确定度分量得到体积VBTB的不确定度u(VBTB).
mlVuT013.
0006.
0012.
0)(22=+=表A2.
2滴定中的数值与不确定度说明数值x标准不确定度u(x)相对标准不确定度u(x)/xrep重复性1.
00.
00050.
0005mBKHPBKHP的重量0.
3888g0.
00013g0.
00033PBKHPBKHP的纯度1.
00.
000290.
00029MBKHPBKHP的摩尔质量204.
2212g/mol0.
0038g/mol0.
000019VBTB滴定KHP用去NaOH的体积18.
64ml0.
013ml0.
0007A2.
5步骤4:合成标准不确定度的计算CBNaOHB由下式计算获得TKHPKHPKHPNaOHVMPmc=1000[mol/l]表A2.
2列出了上述各参数的数值、标准不确定度和相对标准不确定度.
代入上述数值后,得到:10214.
064.
182212.
2040.
13888.
01000=***=NaOHcmol/lCNAS-GL06:2006第59页共148页2006年06月01日发布2006年07月01日实施对于乘法表示式(如上式),按下式使用标准不确定度:lmolccuNaOHNaOH/00010.
000097.
0)(=*=lmolcNaOHcNaOHuccNaOHcNaOHucVVuMMuPPummureprepuccuTTKHPKHPKHPKHPKHPKHPNaOHNaOHc00010.
000097.
0)(00097.
000070.
02000019.
0200029.
0200033.
020005.
02)()()()()()()(22222=*==++++=++++=为简化上式合成标准不确定度的运算,引用了电子表格(见附录E.
2).
表A2.
3为填入适当数值的的电子表格,并加注了解释.
建议检查不同参数的影响大小.
各参数的影响大小可以很直观地用直方图表示.
图A2.
9显示了从表A2.
3计算得到的),(jxyu的大小.
滴定体积TV的不确定度分量是最大的,其次是重复性.
称量过程和滴定标准物的纯度处于同一数量级,而摩尔质量的不确定度却几乎小了一个数量级.
A2.
6步骤5:重新评估显著性不确定度分量V(T)不确定度分量是最大的.
滴定KHP消耗NaOH的体积V(T)受四种量的影响:滴定所消耗体积的重复性、活塞滴定管的校准、滴定管滴定时的温度与滴定管校准时的温度之间差异,以及终点判定的重复性.
对比各分量的大小,校准是最大的.
所以该分量必须研究透彻.
V(T)00.
02u(y,xB1B)(mmolГP1P)图A2.
9标定NaOH的不确定度分量大小M(KHP)P(KHP)m(KHP)重复性c(NaOH)0.
040.
060.
080.
10.
12CNAS-GL06:2006第60页共148页2006年06月01日发布2006年07月01日实施V(T)校准的标准不确定度是由供应商假定为三角形分布计算得到的数据.
分布形状选择的影响见表A2.
4.
根据ISO指南4.
3.
9注1:"对于正态分布,期望值为μ,标准偏差为α,区间μ±3α覆盖了99.
97%的分布.
因此,如果上下限aB+B和aB-B规定了99.
73%界限,而不是100%的界限,则jX可以假定大致为正态分布,而不是对jX(在区间内)所知无几,那么,9/)(22α=jxu.
相对地,半宽度为a的对称矩形分布的方差为3/2a….
.
而半宽度为a的对称三角形分布的方差为6/2a.
相对于这三种分布的假定的差别,这三种分布的方差却处于同一数量级是非常令人惊讶的.
"因此,影响量的分布函数的选择对合成标准不确定度(u())NaOHc数值的影响并不明显,所以将之假定为三角形分布有充分的理由.
扩展不确定度)(NaOHcU可由合成标准不确定度乘以包含因子2后得到.
lmolcUNaOH/0002.
0200010.
0)(=*=所以,NaOH溶液的浓度为(0.
1021±0.
0002)mol/l.
表A2.
3:滴定不确定度的电子表格计算ABCDEFG1Repm(KHP)P(KHP)M(HP)V(T)2数值1.
00.
38881.
0204.
221218.
643不确定度0.
00050.
000130.
000290.
00380.
01345rep1.
01.
00051.
01.
01.
01.
06m(KHP)0.
38880.
38880.
388930.
38880.
38880.
38887P(KHP)1.
01.
01.
01.
000291.
01.
08M(KHP)204.
2212204.
2212204.
2212204.
2212204.
2250204.
22129V(T)18.
6418.
6418.
6418.
6418.
6418.
6531011c(NaOH)0.
1021360.
1021870.
1021700.
1021660.
1021340.
10206512),(ixyu0.
0000510.
0000340.
000030-0.
000002-0.
0000711322),(,)(ixyuyu9.
72E-92.
62E-91.
16E-99E-104E-125.
041E-91415))((NaOHcu0.
000099CNAS-GL06:2006第61页共148页2006年06月01日发布2006年07月01日实施各参数的数值列于第二行C2-G2.
它们的标准不确定度列于下一行C3-G3.
将C2-G2的数据用电子表格程序复制到列B5-B9.
运用上述数据计算的结果(c(NaOH))列于B11.
C5是C2加上它的不确定度C3得到的重复性数值.
由C5-C9计算得到的结果列于C11.
列D和G重复以上步骤得到.
第12行(C12-G12)的数值是行(C11-G11)减去B11的数值得到的.
第13行(C13-G13)是第12行(C12-G12)的平方,相加后得到的数值列入B13.
B15是合成标准不确定度,它等于B13的平方根.
表A2.
4:不同分布假定的影响分布因子u(V(T;cal))(ml)u(V(T))(ml)))(NaOHccu(mol/l)矩形30.
0170.
0190.
00011三角形60.
0120.
0150.
00009正态P(注1)P90.
0100.
0130.
000085注1:因子9来源于ISO导则4.
3.
9注1的因子3(详见48页).
CNAS-GL06:2006第62页共148页2006年06月01日发布2006年07月01日实施例A3:酸碱滴定概要目标以已知浓度的氢氧化钠(NaOH)溶液标定盐酸(HCl)溶液.
测定步骤以邻苯二甲酸氢钾(KHP)标定氢氧化钠(NaOH)溶液,再以氢氧化钠(NaOH)溶液标定盐酸(HCl)的浓度.
其过程如图A3.
1所示.
)()/(),(iiixuxyxyu=的数值取自表A3.
3图A3.
1滴定过程被测量:HClKHPT1T2KHPKHPHClVMVVPm1000c=[moll-1]其中,上式各代号已在表A3.
1中列出.
1000是由ml转化为升的换算系数.
不确定度来源的确定:各相关不确定度来源如图A3.
2所示.
不确定度分量的定量:最终不确定度评估值为0.
00016mol/l.
表A3.
1列出了各数值及其不确定度;图A3.
3以直方图表示了各数值大小.
称取KHP以NaOH滴定KHP图A3.
1滴定过程准确移取以NaOH滴定HCL结果V(HCI)0u(y,xB1B)(mmolГP1P)u(y,xB1B)=(y/xB1B)·u(xB1B)图A3.
3酸碱滴定不确定度分量M(KHP)V(T1)V(T2)P(KHP)m(KHP)复现性с(HCI)0.
050.
10.
150.
2CNAS-GL06:2006第63页共148页2006年06月01日发布2006年07月01日实施表A3.
1酸碱滴定数据及其不确定度名称数值x标准不确定度u(x)相对标准不确定度u(x)/xrep重复性10.
0010.
001KHPmKHP的质量0.
3888g0.
00012g0.
00033KHPPKHP的纯度1.
00.
000290.
000292TV滴定HCl消耗NaOH的体积14.
89ml0.
014ml0.
000941TV滴定KHP消耗NaOH的体积18.
64ml0.
015ml0.
00080KHPMKHP的摩尔质量204.
2212g/mol0.
0038g/mol0.
000019HClV用NaOH滴定移取的HCl的体积15ml0.
011ml0.
00073HClCHCl溶液的浓度0.
10139mol/l0.
00018mol/l0.
0018湿度校准重复性V(T1)P(KHP)c(HCl)m(tare)m(gross)线性线性校准M(KHP)校准图A3.
2酸碱滴定的因果图m(KHP)m(KHP)终点偏差灵敏度灵敏度V(T2)终点偏差湿度校准V(T2)终点V(T2)V(T1)终点V(T1)V(HCI)湿度校准V(HCI)同一部天平CNAS-GL06:2006第64页共148页2006年06月01日发布2006年07月01日实施例A3:酸碱滴定详细讨论A3.
1介绍本例探讨了标定盐酸(HCl)溶液浓度的一系列实验.
另外,也对滴定的技术进行了多方面的探讨.
HCl用刚由邻苯二甲酸氢钾(KHP)标定的氢氧化钠(NaOH)溶液标定.
与例A2一样,假定HCl的浓度在0.
1mol/l这个数量级,滴定终点用自动滴定装置通过pH曲线的形状判断.
上述分析使测量不确定度以SI单位测量术语给出.
A3.
2步骤1:技术规定步骤1详细叙述了测量步骤,包括罗列了一系列的测量步骤和被测量的数学表达式.
步骤测定HCl溶液浓度包括以下各个阶段(可参见图A3.
4)1)干燥滴定标准物邻苯二甲酸氢钾(KHP),以确保其纯度符合其供应商提供的证书上所标数值.
称取大约0.
388g干燥的基准KHP以标定19mlNaOH.
2)将滴定标准物KHP溶解于约50ml的去离子水中,以NaOH滴定.
滴定装置自动地滴加NaOH,同时绘出pH曲线.
通过记录的pH曲线形状确定终点.
3)用移液管移取15mlHCl溶液.
用去离子水稀释至约50ml于滴定瓶中.
4)用同一台自动滴定装置测定HCl溶液的浓度.
称取KHP以NaOH滴定KHP图A3.
4HCI溶液浓度的测定准确移取HCI以NaOH滴定HCL结果CNAS-GL06:2006第65页共148页2006年06月01日发布2006年07月01日实施计算:被测量是HCl溶液的浓度CBHClB.
它取决于KHP的质量、纯度、分子量、两次滴定终点时消耗NaOH的体积和HCl的移取量:HClKHPTTKHPKHPHClVMVVPmc=121000[mollP-1P]其中CBHClB:HCl溶液的浓度[mol/l]1000::由ml转化为l的换算系数mBKHPB::称取的KHP质量[g]PBKHPB:KHP以质量分数表示的纯度VBT2:B滴定HCl所用NaOH溶液的体积VBT1:B滴定KHP所用NaOH溶液的体积MBKHPB:KHP的摩尔质量[g/mol]VBHClB:被NaOH滴定的HCl的体积[ml]A3.
3步骤2:确定和分析不确定度来源用直观的因果图(图A3.
5)表示出各不确定来源及其对被测量的影响是分析测量不确定度分量的最好方法.
重复性评估作为整体可以方法确认研究中得到,因此无需分别考虑所有重复性的分量.
因此这些湿度校准重复性V(T1)P(KHP)c(HCI)m(tare)m(gross)线性线性校准M(KHP)校准m(KHP)m(KHP)终点偏差灵敏度灵敏度V(T2)终点偏差湿度校准V(T2)终点V(T2)V(T1)终点V(T1)V(HCI)湿度校准V(HCI)同一部天平图A3.
5最终因果图CNAS-GL06:2006第66页共148页2006年06月01日发布2006年07月01日实施可以归纳为一种分量(见修订后的因果图A3.
5).
对参数VBT2B、VBT1B、MBKHPB、PBKHPB和MBKHPB的影响因素已在前几个例子中有详尽的探讨,因此只有VBHClB影响量在本节中详细讨论.
U体积VUBUHClUB用移液管移取15ml待测HCl溶液.
从移液管排出的HCl溶液的体积与所有容量测量装置一样有三种同样的不确定度来源:1.
排出体积的变化或重复性2.
移液管所标体积的不确定度3.
移液管移取的溶液温度与校准时温度的差异.
A3.
4步骤3:不确定度分量的量化本步骤的目的就是将步骤2分析的各不确定度来源进行定量.
各分支或不同组分的定量在前两个例子中有详细的描述.
因此,本节只给出不同分量的概要.
U重复性方法确认表明测定的重复性为0.
1%(%rsd).
该数值可以直接用来计算与各重复性有关的合成标准不确定度.
U质量UmBKHPB校准/线性:天平制造商给出了±0.
15mg的线性分量.
该数值代表了托盘上被称量的实际重量与从天平所读取的数值的最大差值.
线性分量被假设成矩形分布,换算成标准不确定度为:mg087.
0315.
0=线性分量应重复计算两次,一次是空盘,另一次为毛重,产生的不确定度u(mBKHPB)为:2)087.
0(2)(*=KHPmu=0.
12mg注1:由于对非线性的形式未做任何假设,因此将该分量重复计算了两次.
非线性被相应地看作为对每次称重的系统影响,在称量范围内影响的大小是随机变化的.
注2:因为所有称重均是以常规方式在空气中完成的,因此未考虑浮力修正[H.
19].
余下的不确定度太小,可以忽略不计.
参见附录G中的注1.
UP(KHP)供应商证书上给出的P(HKP)值为100%±0.
05%,其引用的不确定度可考虑为矩形分布,标准不确CNAS-GL06:2006第67页共148页2006年06月01日发布2006年07月01日实施定度为:00029.
030005.
0)(==puKHPUV(T2)ⅰ)校准:制造商提供的数值(±0.
03ml),近似于三角形分布0.
03/6=0.
012mlⅱ)温度:温度变化的范围为±4℃,近似于矩形分布15*2.
1*10P-4P*4/3=0.
007mlⅲ)终点判定偏差:在氩气中滴定可以消除空气中的COB2B造成判定的终点与等当点的偏差.
不确定度可不予考虑.
VBT2B求得为14.
89ml,将两个分量合成为体积VBT2B的不确定度μ(VBT2B):mlVuT014.
0007.
0012.
0)(222=+=U体积VUBUT1UB除温度外,所有分量均与VBT2B相同.
ⅰ)校准:0.
03/6=0.
012mlⅱ)温度:滴定0.
3888gKHP大约消耗NaOH的体积为19ml,因此其不确定度分量为19*2.
1*10P-4P*4/3=0.
009mlⅲ)偏差:可忽略VBT1B求得为18.
64ml,其标准不确定度u(VBT1B):mlVuT015.
0009.
012.
0)(221=+=U摩尔质量MUBKHPBKHP(CB8BHB5BOB4BK)各组成元素的原子量及其不确定度(从最新的IUPAC原子量表查得)为:元素原子量不确定度标准不确定度C12.
0107±0.
00080.
00046H1.
00794±0.
000070.
000040O15.
9994±0.
00030.
00017K39.
0983±0.
00010.
000058对于每个元素来说,其标准不确定度可按IUPAC给出的数值以矩形分布求得.
将所给的数值除以3可得到其标准不确定度.
CNAS-GL06:2006第68页共148页2006年06月01日发布2006年07月01日实施KHP摩尔质量MBKHPB及其不确定度分别为:MBKHPB=8*12.
0107+5*1.
00794+4*15.
9994+39.
0983=204.
2212g/molu(MBKHPB)=2222000058.
0)00017.
04()00004.
05()00046.
08(+*+*+*=0.
0038g/mol注:单个原子的不确定度分量并非独立的.
因此计算原子分量的不确定度时将原子量的标准不确定度乘以原子数.
U体积VUBUHClUBⅰ)校准:制造商给定15ml移液管的不确定度为±0.
02ml,近似为三角形分布0.
02/6=0.
008mlⅱ)温度:实验室温度变化介于±4℃之间.
采用矩形分布,其标准不确定度为:15*2.
1*10P-4P*4/3=0.
007ml合成上述不确定度分量:u(VBHClB)=222007.
0008.
00037.
0++=0.
011ml表A3.
2酸碱滴定数据及其不确定度(两次滴定过程)名称数值x标准不确定度u(x)相对标准不确定u(x)/xrep重复性10.
0010.
001KHPmKHP的质量0.
3888g0.
00012g0.
00031KHPPKHP的纯度1.
00.
000290.
000292TV滴定HCl用去NaOH的体积14.
89ml0.
014ml0.
000941TV滴定KHP用去NaOH的体积18.
64ml0.
015ml0.
00080KHPMKHP的摩尔质量204.
2212g/mol0.
0038g/mol0.
000019HClV滴定NaOH移取HCl的体积15ml0.
011ml0.
00073A3.
5步骤4:计算合成标准不确定度cBHClB由下式计算得到:HClKHPTTKHPKHPHClVMVVPmc=121000注:本例中,重复性评估视为相对影响;因此完整的公式应为CNAS-GL06:2006第69页共148页2006年06月01日发布2006年07月01日实施repVMVVPmcHClKHPTTKHPKHPHCl*=121000先后两次滴定实验的所有中间值及其标准不确定度均列于表A3.
2中.
代入这些数值后110139.
01152212.
20464.
1889.
140.
13888.
01000=******=mollcHCl相应地合成各不确定度分量:lmolcHClcHClucrepuVVuMMuVVuVVuPPummuccuHClHClKHPKHPTTTTKHPKHPKHPKHPHClHCl00018.
00018.
0)(0018.
0001.
000073.
0000019.
000080.
000094.
000029.
000031.
0)()()()()()()()(222222222221122222=*==++++++=++++++=电子表格方法(见附录E)可用于简化上述合成不确定度的计算.
表A3.
3中的电子表格已填入了相应数值,并附有相应的解释.
不同不确定度分量的大小可以用直方图的形式比较.
图A3.
6将表A3.
3中各分量),(jxyu的值用直方图表示.
将合成标准不确定度乘以包含因子2计算扩展不确定度U(cBHClB):U(cBHClB)=0.
00018*2=0.
0004mol/lHCl溶液的浓度为:(0.
1014±0.
0004)mol/lV(HCI)0u(y,xB1B)(mmolГP1P)图A3.
6酸碱滴定的不确定度M(KHP)V(T1)V(T2)P(KHP)m(KHP)重复性с(HCI)0.
050.
10.
150.
2CNAS-GL06:2006第70页共148页2006年06月01日发布2006年07月01日实施表A3.
3酸碱滴定----电子表格计算不确定度ABCDEFGHI1repm(KHP)P(KHP)V(T2)V(T1)M(KHP)V(HCl)2数值1.
00.
38881.
014.
8918.
64204.
2212153不确定度0.
0010.
000120.
000290.
00140.
0150.
00380.
01145rep1.
01.
0011.
01.
01.
01.
01.
01.
06m(KHP)0.
38880.
38880.
388920.
38880.
38880.
38880.
38880.
38887P(KHP)1.
01.
01.
01.
000291.
01.
01.
01.
08V(T2)14.
8914.
8914.
8914.
8914.
90414.
8914.
8914.
899V(T1)18.
6418.
6418.
6418.
6418.
6418.
65518.
6418.
6410M(KHP)204.
2212204.
2212204.
2212204.
2212204.
2212204.
2212204.
2250204.
221211V(HCl)1515151515151515.
0111213c(HCl)0.
1013870.
1014890.
1014180.
1014170.
1014820.
10130601013850.
10131314),(xyu0.
0001010.
0000310.
0000290.
000095-0.
000082-0.
0000019-0.
0000741522),(,)(ixyuyu3.
34E-81.
03E-89.
79E-108.
64E-109.
09E-96.
65E-93.
56E-125.
52E-91617))((HClcu0.
00018各参数的数值列于第二行C2到I2.
它们的标准不确定度列于下一行(C3-I3).
用电子表格将C2-I2的数值复制到第二列从B5到B11.
利用上述数据计算得到的结果(c(HCl))列于B13.
C5显示的是重复性的数值,它等于C2加上其不确定度C3.
将C5-C11的数据计算的结果列于C13.
列D至列I重复类似的过程.
第14行(C14-I14)的数值是行(C13-H13)减去B13的结果.
第15行(C15-I15)是第14行(C14-I14)的平方,其平方和列于B15.
B17给出了合成标准不确定度,等于B15的平方根.
A3.
6滴定例子中的特殊性滴定实验的特殊性在本例第二部分中进行探讨.
研究实验准备或滴定过程中哪些因素的变化会对最终结果产生影响以及其不确定度很有趣的事.
U25℃平均室温的影响对于常规分析,分析化学师很少会对实验室温度对体积的系统影响进行修正.
这种情况涉及到修CNAS-GL06:2006第71页共148页2006年06月01日发布2006年07月01日实施正所引入的不确定度.
体积测量装置是在20℃条件下校准的.
但是很少有实验室配备了温度控制装置来保持在该温度.
为了说明,考虑对25℃的平均室温为进行修正.
应使用修正后的体积来计算最终分析结果,而不是按20℃时校准的体积计算.
按照下式,修正温度对体积的影响:)]20(1['=TVVα其中V':平均温度T时的实际体积V:20℃时的校准体积α:水溶液的膨胀系数[℃P-1P]T:实验室实际观测到的温度[℃]被测量的计算公式应重写为:''1'21000HClTTKHPKHPKHPHClVVVMPmc=加入温度修正项后:()[]()[]()[]*==2012012011000100012''1'2TVTVTVMPmVVVMPPmHClcHClTTKHPKHPKHPHClTTKHPKHKHPααα上式在假设温度T和水溶液的膨胀系数α对于三个体积均是相同的,可以简化为:*=)]20(1[100012TVVVMPmcHCITTKHPKHPKHPHCIα上式给出的HCl结果与20℃时称有差别:1410149.
0)]2025(101.
21[1564.
182236.
20489.
140.
13888.
01000=*******=mollcHCI该数值仍然落在平均温度为20℃时结果的合成标准不确定度给出的范围内,说明结果无显著影响.
由于25℃平均室温的温差变化假设为±4℃,因此,温度的变化并不影响合成标准不确定度的评价.
U肉眼判断滴定终点CNAS-GL06:2006第72页共148页2006年06月01日发布2006年07月01日实施如果用酚酞代替由pH曲线求等当点的自动终点识别装置作终点判断,就会引入误差.
由透明向红/粉红色转变的pH值介于8.
2至9.
8之间,这会额外增加滴定量,相对测定pH值的自动终点识别装置来说引入了误差.
研究显示这会使体积增加0.
05ml,同时肉眼判断的标准不确定度大约为0.
03ml.
额外增加的体积必须在最终的结果中考虑到.
肉眼判断时实际的体积为:ExcessTIndTVVV+=1;1IndTV;1:肉眼判断终点时的体积1TV:等当点时的体积ExcessV:使酚酞变色需要增加的体积上述体积的修正导致被测量计算式的改变:HCIExcessIndTKHPExcessIndTKHPKHPHCIVVVMVVPmc=)()(1000;1;2标准不确定度u(VBT2B)和u(VBT1B)必须用肉眼判断终点的标准不确定度作为终点判断重复性的不确定度分量重新计算,mlVVuVuExcessIndTT034.
003.
0009.
0012.
0004.
0)()(2222;11=+++==mlVVuVuExcessIndTT033.
003.
0007.
0012.
0004.
0)()(2222;22=+++==合成标准不确定度)(HClccu=0.
0003mol/l比以前大得多.
U三次测定求得最终结果两次滴定实验重复了三次后,求得最终结果.
三次测定可减少重复性的分量,减少总不确定度.
正如本例第一部分所述,所有重复实验的变化都合并成一个组分,作为总的实验重复性,见图A3.
5所示的因果关系图.
各不确定度分量通过下列方式计算:U质量UmBKHPB线性:0.
15/mg087.
03=mgmuKHP12.
087.
02)(2=*=U纯度UPBKHPB纯度:00029.
03/005.
0=CNAS-GL06:2006第73页共148页2006年06月01日发布2006年07月01日实施U体积VUBUT2UB计量:ml012.
06/03.
0=温度:ml007.
03/4101.
2154=***mlVuT014.
0007.
0012.
0)(222=+=U重复性三次测定的质量记录显示长期的实验平均标准偏差为0.
001(以RSD表示).
不建议使用三次测定的实际标准偏差,因为它本身包含了52%的不确定度.
三次测定(次独立测定)的标准不确定度由0.
001除以3得到:00058.
03/001.
0Re==p(RSD)U体积UHClV校准:ml008.
06/02.
0=温度:ml007.
03/4101.
2154=***mlVuKHP01.
0007.
0008.
0)(22=+=U摩尔质量MUBUKHPUBU:0038.
0)(=KHPMug/molU体积U1TV校准:ml02.
06/03.
0=温度:ml009.
03/4101.
2194=***mlVuT015.
0009.
0012.
0)(221=+=所有不确定度分量的值均收于表A3.
4.
合成标准不确定度为0.
00016mol/l,由于是三次测定因此是略有下降.
各不确定度分量的直方图图示对比见图A3.
7,可明显看出那个结果的一些理由.
虽然重复性分量大大降低了,体积的不确定分量仍保持原来的数值,限制了进一步的改善.
V(HCI)0u(y,xB1B)(mmolГP1P)图A3.
7重复实验酸碱滴定的值和不确定度M(KHP)V(T1)V(T2)P(KHP)m(KHP)复现性с(HCI)0.
050.
10.
150.
2重复实验单次实验CNAS-GL06:2006第74页共148页2006年06月01日发布2006年07月01日实施表A3.
4重复酸碱滴定的数值和不确定度名称数值x标准不确定度u(x)相对标准不确定u(x)/xrep重复性1.
00.
000580.
00058KHPmKHP的质量0.
3888g0.
00013g0.
00033KHPPKHP的纯度1.
00.
000290.
000292TV滴定HCl用去NaOH的体积14.
90ml0.
014ml0.
000941TV滴定KHP用去NaOH的体积18.
65ml0.
015ml0.
0008KHPMKHP的摩尔质量204.
2212g/mol0.
0038g/mol0.
000019HClV滴定NaOH移取出HCl的体积15ml0.
01ml0.
00067CNAS-GL06:2006第75页共148页2006年06月01日发布2006年07月01日实施例A4实验室内部确认研究的不确定度评估面包中有机磷农药的测定概要目的使用萃取和GC方法测定面包中残留的有机磷农药的含量.
测定程序测定面包中残留的有机磷农药所需的步骤见图A4.
1.
被测量16hom10Re=mgkgFmcIVCIPsamplerefoprefopop其中:opp:样品中农药的含量水平][1mgkgopI:样品萃取液的峰响应值refC:参考标准的质量浓度][1gmlμopV:萃取样品的最终体积[ml]610:从][1gg到[1mgkg]的换算因子refI:参考标准的峰响应值Rec:回收率samplem:待测子样品的质量[g]homF:样品非均匀性校正因子确定不确定度来源相关不确定度来源见图A4.
2的因果图.
不确定度分量的量化基于实验室内部确认数据,三个主要的分量见表A4.
1,直方图表示见图A4.
3(数值取自表A4.
5).
混合均匀萃取图A4.
1有机磷农药分析过滤稀释定容GC测定制备校准标准GC校准结果CNAS-GL06:2006第76页共148页2006年06月01日发布2006年07月01日实施表A4.
1:农药分析的不确定度描述数值x标准不确定度u(x)相对标准不确定度u(x)/x评述重复性(1)1.
00.
270.
27基于不同类型样品的平行试验偏差(回收率)(2)0.
90.
0430.
048加料样品其它来源(3)(均匀性)1.
00.
20.
2基于假设模型的评估u(PBopB)/PBopB----0.
34相对标准不确定度F(hom)P(op)图A4.
2农药分析的不确定度来源重复性m(样品)V(op)稀释V(ref)m(ref)l(ref)l(op)校准t(op)V(ref)m(ref)稀释校准温度校准校准线性c(ref)纯度(ref)V(op)V(op)校准温度回收率l(ref)校准m(样品)m(gross)校准线性m(tare)校准线性均匀性偏差重复性P(op)00.
10.
20.
30.
4u(y,xB1B)(mgГP1P)u(y,xB1B)=(y/xB1B)·u(xB1B)的数值取自表A4.
5图A4.
3农药分析的不确定度CNAS-GL06:2006第77页共148页2006年06月01日发布2006年07月01日实施混合均匀萃取图A4.
4有机磷农药分析过滤稀释定容GC测定制备校准标准GC校准结果例A4面包中有机磷农药的测定详细讨论A4.
1介绍这个例子说明了用实验室内部确认的数据计算测量不确定度的方法.
本测量的目的是测定面包中残余有机磷农药的含量.
通过测定加料样品,实验室确认计划和实验建立起溯源性.
假设对样品中所加的料和被分析物的测量响应的不同而导致的不确定度相对于结果的总不确定度是小的.
A4.
2步骤1:技术规定有关被测量更详细分析方法的技术规定最好是综合描述分析方法的不同的步骤,并提供被测量的计算公式.
程序:测定程序在图A4.
4中以图的方式说明.
各步骤如下:ⅰ)均匀性:将完整的样品切成小碎片(约2cm),从中随机选择15个小碎片,并把该子样品混合均匀.
当观察到极度不均匀性时,在混合前按比例取样.
ⅱ)称量分析用的子样品,给出samplemⅲ)萃取:用有机溶剂定量萃取被分析物,转移并通过硫酸钠的柱子进行脱水,用Kuderna-Danish装置浓缩萃取液.
ⅳ)液-液萃取:ⅴ)乙腈/己烷分配液,用己烷洗乙腈萃取液,通过硫酸钠柱干燥已烷层.
ⅵ)浓缩洗过的萃取液,用气体把萃取液吹至近干.
ⅶ)稀释至标准体积opV(约2ml),用10ml刻度管.
ⅷ)测定:注射5μl样品萃取液至GC中,得到峰响应值opI.
ⅸ)制备约1.
5mlgμ标准溶液(实际质量浓度为refC).
ⅹ)用制备的标准溶液作GC校准.
注射5l标准溶液到GC中,得到参考标准峰响应值refI.
计算:CNAS-GL06:2006第78页共148页2006年06月01日发布2006年07月01日实施最终样品萃取液的质量浓度opc为:1=gmlIIccrefoprefopμ散样中的农药含量PBopB估计值为:1610Re=mgkgmcVcPsampleopopop或将opC带入得:1610Re=mgkgmcIVcIPsamplerefoprefopop其中:opp:样品中农药的含量水平[1mgkg]opI:样品萃取液的峰响应值refC:参考标准的质量浓度[1gmlμ]opV:萃取液的最终体积[ml]610:从[1gg]到[1mgkg]的换算因子refI:参考标准的峰响应值Rec:回收率samplem:待测子样品的质量[g]范围:此分析方法适用于小范围的化学性质相近,含量介于0.
01至2mg1kg之间的各类的面包基体中的农药.
A4.
3步骤2识别和分析不确定度来源对这样一个复杂的分析程序,识别所有的不确定度来源最好方法是画一个因果图.
被测量计算公式中的参数在图作为主要分支.
考虑到分析程序(A4.
2)的每一步,进一步将影响因素添加到该图上,直到所有重要贡献因素均予充分考虑.
CNAS-GL06:2006第79页共148页2006年06月01日发布2006年07月01日实施样品的非均匀性不是原始被测量公式中的一个参数,但是它在分析程序中对分析结果有很大的影响.
因此在因果图中加上了一个新的分支,(hom)F,代表样品的非均匀性(图A4.
5).
最后,样品非均匀性引入的不确定度分量应该包括进被测量的计算公式.
为了明确地表明非均匀性引起的不确定度的影响,被测量计算公式应写成下式是有益的:][1016hom=mgkgmRIVcIFPsampleecrefoprefopop其中homF是校正因子,在原先计算公式中设为1.
这就明确了校正因子的不确定度必须包括在总的不确定度的评价中.
这个最终的公式表明了不确定度是怎样应用的.
注:校正因子:这个方法非常概括,可能对突出那些隐藏着的假设很有用.
原则上,每次测量都与这些通常设为1的校正因子联系在一起.
例如,opc的不确定度可以被表示为opc的标准不确定度,或是表示为校正因子不确定度的标准不确定度.
对于后者,该数值等同于以相对标准偏差表示的opc的不确定度.
A4.
4步骤3:不确定度分量的量化根据第7.
7节,不同的不确定度分量的量化可以利用实验室内部开发和确认研究中的数据.
·分析过程随机变化总体的现有的最佳估计值·总体偏差(回收率)及其不确定度可能的最佳估计值F(hom)P(op)图A4.
5将样品非均匀性作为一个主要分支后的因果图校准l(op)V(ref)m(ref)稀释校准温度校准校准线性c(ref)纯度(ref)V(op)V(op)校准温度回收率I(ref)校准m(样品)m(gross)校准线性m(tare)校准线性精密度精密度精密度精密度精密度精密度精密度灵敏度灵敏度精密度CNAS-GL06:2006第80页共148页2006年06月01日发布2006年07月01日实施·总体性能研究不能全面考虑的其他影响因素有关的不确定度的量化为使这些输入数据的关系和范围更加明确,有必要对因果图做些重新安排(图A4.
6).
注:通常,测量用的样品按小批量来管理,每一批量包括一个校准样,控制偏差的回收率检查样品,以及检查精密度的随机平行样.
假如这些检查表明显著地偏离确认研究过程中所确定的性能,应采取修正措施.
这些基本的质量控制满足在日常测试不确定度评估中使用确认方法研究的数据的主要要求.
在因果图中插入额外影响因子"重复性",计算opP的模型变为:psampleecrefoprefopopFmRIVcIFPRe6hom10=1mgkg公式A4.
1这就是说,象对待均匀性一样,重复性用一个乘法因子pFRe来表示.
选择这样的形式是为了计算方便,就象以下所述.
现在考虑不同的影响因素.
1.
U精密度研究对不同面包样品中的典型有机磷农药进行一系列平行测试(同一均匀样品、完整的萃取/测定程序),以获得该分析程序的总的随机变化(精密度).
测定结果见表A4.
2.
标准化的差值数据(差值除以平均值)提供了总的随机变化的量度.
为了得到单次测量的相对标准不确定度的估计值,求该标准化差值的标准偏差并除以2,这样就可以把成对差值的标准偏差校F(hom)图A4.
6使用方法确认研究的数据后重新安排的因果图重复性m(样品)V(op)稀释V(ref)m(ref)l(ref)l(op)校准t(op)V(ref)m(ref)稀释校准温度校准校准线性c(ref)纯度(ref)V(op)V(op)校准湿度回收率l(ref)校准m(样品)m(gross)校准线性校准线性P(op)CNAS-GL06:2006第81页共148页2006年06月01日发布2006年07月01日实施正为单次测定值的标准不确定度.
这就给出了总的测试程序的随机变化的标准不确定度,包括回收率的随机变化,但不包括样品非均匀性影响,此标准不确定度为:27.
02/382.
0=.
注:初一看,平行测试提供的自由度可能不够.
但是做以上测试并不是为了得到特定种类的面包表A4.
2:农药分析平行测试的结果农药残留D1(mgkgP-1P)D2(mgkgP-1P)平均值(mgkgP-1P)D1与D2的差值差值/平均值马拉硫磷1.
301.
301.
300.
000.
000马拉硫磷1.
300.
901.
100.
400.
364马拉硫磷0.
570.
530.
550.
040.
073马拉硫磷0.
160.
260.
21-0.
10-0.
476马拉硫磷0.
650.
580.
620.
070.
114PirimiphosMethyl0.
040.
040.
040.
000.
000甲基毒死蜱0.
080.
090.
085-0.
01-0.
118PirimiphosMethyl0.
020.
020.
020.
000.
000甲基毒死蜱0.
010.
020.
015-0.
01-0.
667PirimiphosMethyl0.
020.
010.
0150.
010.
667甲基毒死蜱0.
030.
020.
0250.
010.
400甲基毒死蜱0.
040.
060.
05-0.
02-0.
400PirimiphosMethyl0.
070.
080.
75-0.
01-0.
133甲基毒死蜱0.
010.
010.
100.
000.
000PirimiphosMethyl0.
060.
030.
0450.
030.
667中一种特定农药的分析测试程序的精密度的准确数据.
以上测试更重要的是测试一系列不同的物质和不同的样品中所选择的有代表性的典型有机磷农药的含量.
可通过对许多材料的平行测试这种最有效的方法来达到,该方法(重复性估计)中每一种材料平行测试的自由度约为1.
2.
U偏差研究分析程序的偏差是通过在实验室内部确认研究中使用加料样品(将均匀的样品分份,其中的1份加料)来进行研究的.
表A4.
3收集了各种类型加料样品样品的长期研究的结果.
相关的线框填入有关的"面包"类型,显示了42个样品的平均回收率为90%,标准偏差为(s)为28%.
标准不确定度采用平均值的标准偏差:0432.
042/28.
0)eR(==cu显著性检测是用来确定平均回收率是否与1.
0有显著性差异.
检测统计数据t用下式来计算:CNAS-GL06:2006第82页共148页2006年06月01日发布2006年07月01日实施315.
20432.
0)9.
01()eR(eR1===cuct这个数据与95%置信度,n-1自由度的双边临界值critt比较(其中n是用来评估ceR的测试结果的数目),假如t大于或等于critt值,则ceR与1有显著性差异.
021.
231.
241,≥=crittt在这个例子中,使用了校正因子(ceR1),因此ceR被明确地包括在结果的计算中.
表A4.
3:农药残留的回收率研究物质残留类型浓度(mgkgP-1P)NP1)P平均值P2)P(%)SP2)P(%)废油PCB10.
08849黄油OC0.
653310912动物饲料1OC0.
325100909动物和蔬菜油脂1OC0.
3334102241987芸苔OC0.
323210418面包OP0.
13429028甜面包干OP0.
13308427肉和骨饲料OC0.
32589512玉米麸质OC0.
3259929油菜饲料1OC0.
325118913小麦饲料1OC0.
32525889黄豆饲料1OC0.
325138519大麦饲料1OC0.
32598422(1)测试次数.
(2)平均值和样品标准偏差s以回收率百分比给出.
3.
U其它不确定度来源图A4.
7的因果图表明哪一个其它不确定度来源是:(1)充分地地包括在精密度数据中;(2)包括在回收率中;(3)必须进一步研究,并最终在不确定度计算中加以考虑.
所有的天平和重要的体积测量装置处于常规的控制下.
精密度和回收率研究已经考虑了体积测量装置校准的影响,因为在研究中使用不同的容量瓶和移液管.
持续超过了半年时间的大量的随机变化研究,也包括了环境温度对结果的影响.
因此只剩下参考物质的纯度、GC响应中可能的非线性(在图CNAS-GL06:2006第83页共148页2006年06月01日发布2006年07月01日实施中用校准项refI和opI表示)和样品的均匀性这些不确定度分量需要研究.
制造商提供的标准物质的纯度为99.
53%±0.
06%.
纯度是潜在的不确定度来源,其标准不确定度为00035.
03/0006.
0=(矩形分布).
但是这个分量的贡献如此之小(如与精密度评估相比),所以很明显可以忽略这个分量.
在给定的浓度范围内有关的有机磷农药的响应线性已在方法确认研究中加以确认.
此外,在表A4.
2和表A4.
3中所给类型的不同含量测试研究中,非线性对观察到的精密度有贡献,因此不需要额外考虑.
实验室内部确认方法研究已经证明了这一点.
面包子样品的均匀性是最后剩下的不确定度来源.
尽管已经进行了大规模的文献检索,但还是没有有关面包产品中痕量有机成分分布的文献数据.
(初一看,这很令人惊讶,但大多数分析家总是尽量使样品均匀,而不愿意单独对非均匀性做出评估),而且直接测定均匀性也是不实际的.
因此可以根据使用的取样方法来评估这个不确定分量.
为了有助于评估,考虑了一系列可能的的农药残留分布.
并使用简单的二项式统计分布来计算被分析样品中农药总量的标准不确定度(见A4.
6节).
农药残留分布和最终样品中农药含量的相对标准不确定度计算结果如下:·农药残留仅分布在样品最表面:0.
58·农药残留均匀地分布在样品表面:0.
20·农药残留均匀地分布在整个样品中,但是因为蒸发和分解的原因,越靠近表面农药残留的浓度越低:0.
05-0.
10(取决于"表面层"的厚度).
分布(a)特别适于比例取样或者是完全均匀的:装饰性添加物(全谷物)加到一个表面上会是这种分布.
分布(b)被认为可能是最差的情况,分布(c)被认为最有可能,但是不易与(b)区别.
基于此,选择0.
20这个数值.
注:非均匀性模型的更详细的细节见本例的最后一节.
A4.
5步骤4:;计算合成标准不确定度在分析程序的实验室内部确认研究中,重复性、偏差和所有其它可能的不确定度来源已被彻底研究.
它们的数值和不确定度收集在表A4.
4中.
因为在数学模型(等式A4.
1)中都是相乘关系,因此这些数值可以被合成为:opopcopopcPPuPPu*==++=34.
0)(34.
02.
0048.
027.
0)(222CNAS-GL06:2006第84页共148页2006年06月01日发布2006年07月01日实施本例的电子表格形式(表A4.
5)见4.
5.
注意电子表格计算的是标称修正结果1.
1111的绝对值的不确定度(0.
373),给出的值是相对标准不确定度0.
373/1.
111=0.
34三个不同的不确定度分量的相对大小可用直方图来比较,图A4.
8所示的值),(ixyu取自表A4.
5.
重复性是测量不确定度的最大分量.
由于这个分量是从方法总的变异性得出的,可以进一步实验查明哪些可以改进.
例如,在取样前均匀一整条面包可以显著地降低不确定度.
扩展不确定度U(PBopB)是合成不确定度乘以包含因子2得到:opopoppppU*=**=68.
0234.
0)((1)重复性在分析程序的变异性研究中加以考虑(公式A4.
1中的FBRepB).
(2)在分析程序的偏差研究中加以考虑.
(3)在其它来源的不确定度评价中加以考虑.
A4.
6特殊部分:建立有机磷农药不确定度的非均匀性模型假设样品中所有有关的物质,无论其状态如何,均可以萃取出来进行分析,非均匀性最糟糕的情况是所有待测的物质全部集中在样品的一个或几个部分.
一个常见却紧密联系例子是在整个样品中的不同的部分有两个含量的待测物质,如LB1B和LB2B.
如果是随机抽样,这类非均匀性的影响可使用二项式统计法评估.
所需的数值是:在切开样品后随机选取的n个等份样品中待测物质含量的平均值μ和标准偏差σ.
表A4.
4:农药分析的不确定度F(hom)(3)图A4.
7不确定度其它来源的评估重复性(1)m(样品)V(op)稀释V(ref)m(ref)l(ref)l(op)校准(3)l(op)V(ref)m(ref)稀释校准(2)温度(2)校准(2)线性c(ref)纯度(ref)V(op)V(op)回收率(2)l(ref)校准(3)m(样品)m(gross)线性线性校准(2)校准(2)温度(2)校准(2)校准(2)CNAS-GL06:2006第85页共148页2006年06月01日发布2006年07月01日实施描述数值x标准不确定度u(x)相对标准不确定度u(x)/x评述重复性(1)1.
00.
270.
27不同类型样品的平行测试偏差(回收率)(2)0.
90.
0430.
048加料样品其它来源(3)(均匀性)1.
00.
20.
2基于假设模型的评估U(PBopB)/PBopB0.
34相对标准不确定度表A4.
5:农药分析的不确定度ABCDE1重复性偏差均匀性2数值1.
00.
91.
03不确定度0.
270.
0430.
245重复性1.
01.
271.
01.
06偏差0.
90.
90.
9430.
97均匀性1.
01.
01.
01.
289PBopB1.
11111.
41111.
06041.
33310u(y,xBiB)0.
30-0.
05070.
22211u(y)P2P,u(y,xBiB)P2P0.
14200.
090.
002570.
049381213u(PBopB)0.
377(0.
377/1.
111=0.
34,相对标准不确定度)各个参数的值输入到第二行C2到E2.
它们的标准不确定度在其下行(C3:E3).
电子表格把C2-E2的数据复制到均匀性偏差重复性P(op)00.
10.
20.
30.
4u(y,xB1B)(mgГP1P)u(y,xB1B)=(y/xB1B)·u(xB1B)的数值取自表A4.
5图A4.
8农药分析的不确定度CNAS-GL06:2006第86页共148页2006年06月01日发布2006年07月01日实施第二列B5到B7.
使用这些值计算出的值在B9(=B5*B7/B6,基于公式A4.
1).
C5是C2加上C3其不确定度后的重复性的值.
使用C5:C7的数据计算得到的结果在C9.
在D和E列遵循相同的程序.
第10行(C10:E10)的数据是第9行(C9:E9)的数据减去B9得到的.
第11行数据(C11:E11)是第10行(C10:E10)的数据平方,把(C11:E11)相加得到B11.
B11平方根得到B13,这是合成不确定度.
这些数值如下:22112211)()(nlllnplplpn+=+=μμ[1]221112)()1(llpnp=σ[2]其中1l和2l是从整个样品中总量为X含总体分数分别为1L和2L区域中抽取样品中待测物质的含量;1p和2p是从那些区域中选择含有等份样品的概率(与可选择的总体等份样品数量相比,n必须要小).
以上的数值可用以下的方式计算,假设一个典型的面包样品,大约12*12*24cm,每一小等份样品的尺寸为2*2*2cm(总共有432个等份样品),假设从其中随机选择15个等份样品并均匀.
例(a)待测物质限于样品的大的单个表面(顶表面).
因此2L是0,2l也是0;1L是1,每一份含有顶表面的等份样品就含有物质含量1l,根据给出的尺寸,显然等份样品的6分之1(2/12)满足该标准,因此1p是1/6或0.
167,1l是x/72(也就是有72份"顶表面"等份样品),可以给出:58.
044.
108.
208.
2)17.
01(167.
0155.
2167.
0151212121211=====***==**=μσσσμRSDllllll注:为了计算在整个样品中的含量X,乘以432/15,得到x的平均值的估计值:xxlx=*=**=72725.
2154321这个结果是典型的随机取样;平均值的期望值正好等于总体的平均值.
对于随机取样,除了已在这里表示为σ或RSD的重复性变化外,对总的不确定度没有其他贡献.
例(b)物质均匀地分布在所有平面上.
遵循以上相似的讨论,并假设所有的表面等份样品包含相同的物质含量21,ll也是0,1p根据所给的尺寸计算如下:CNAS-GL06:2006第87页共148页2006年06月01日发布2006年07月01日实施63.
0)241212()2088()241212(1=******=p也就是1p是"外部"2cm样品的分数.
使用相同的假设,则2721xl=注:与例(a)相比数值有变化.
2.
087.
15.
35.
3)63.
01(63.
0155.
963.
01512221211===*==***==**=μσσσμRSDllllll例(c)由于蒸发或其它损失,物质含量在接近表面时减少至0.
这个例子可以简单地考虑为例(b)的反面,这样160,37.
011xlp==,这样:33.
087.
15.
35.
3)37.
01(37.
0156.
537.
0151212121211===*==***==**=μσσσμRSDllllll可是,假如损失延伸的深度小于所移取的等份样品的尺寸如所估计的,每小块都含有1l和2l,1l和2l都不为零.
例如,所有外部等份样品均含有样品的50%的"中心"部分和50%的"外部"部分.
2962121xlll=*=2222122222215.
3)()37.
01(37.
0156.
201537.
01515)(37.
015lllllllll=***==*+**=*+**=σμ给出RSD是:1.
87/20.
6=0.
09在当前模型中,待测物质损失的厚度为1cm的,考查典型的面包样品其面包皮厚度小于或等于1cm,把这作为待测物质损失的深度,(面包皮的形成抑制了该深度下待测物质的损失),据此例(c)的实际变化,将给出所得的值σ/μ不超过0.
09.
注:在本例中,因为非均匀性比抽取作均匀化的等份样品要小,不确定度的降低就多.
一般来说,这将导致对不确定度的减少.
据此,当较大量的内含物(例如含在面包块中颗粒)中含有不成比例的CNAS-GL06:2006第88页共148页2006年06月01日发布2006年07月01日实施待测物质的量时,就无需另外建立模型.
假如含在抽取作均匀化的等份样品中的此类内含物的概率足够大,则其对不确定度的贡献将不会超过上述各例已计算的值.
CNAS-GL06:2006第89页共148页2006年06月01日发布2006年07月01日实施例子A5:原子吸收光谱法测定陶瓷中镉溶出量概要目的用原子吸收光谱法测定陶瓷器皿中镉溶出量,使用的测量程序是经验方法BS6748.
测量程序图A5.
1的流程图列出了测定陶瓷器皿中镉溶出量的各个步骤.
被测量dmmg2temptimeacidVL0fffdaVcr=表A5.
1描述了各变量.
不确定度来源的识别有关的不确定度来源如图A5.
2的因果图所示.
不确定度来源的量化不同分量的大小列于表A5.
1中,分量之间的比较见图A5.
2中的直方图.
图A5.
1可浸取金属步骤表A5.
1:测定镉溶出量的不确定度描述值x标准不确定度u(x)相对标准不确定度u(x)/xCB0B浸取液中镉含量0.
26mglP-1P0.
018mglP-1P0.
069d稀释系数(如使用的话)1.
0P注1P0P注1P0P注1PVBLB浸取液体积0.
332l0.
0018l0.
0054aBVB容器的表面积2.
37dmP2P0.
06dmP2P0.
025fBacidB酸浓度的影响1.
00.
00080.
0008fBtimeB浸泡时间的影响1.
00.
0010.
001fBtempB温度的影响1.
00.
060.
06r每单位面积镉溶出量0.
036mgdmP-2P0.
0033mgdmP-2P0.
09注1:本例中没有用到稀释.
因此d正好为1.
0.
准备表面处理用4%V/V醋酸填充样品浸泡均匀搅拌浸泡液AAS测定AAS校准结果制备校准标准CNAS-GL06:2006第90页共148页2006年06月01日发布2006年07月01日实施图A5.
2测定浸出镉的不确定度来源图A5.
3测定浸出镉的不确定度校准曲线f(酸)f(时间)f(温度)填充温度校准读数结果r1长度2长度面积c(0)V(L)a(V)df(temp)f(time)f(tacid)a(V)V(L)c(O)r12034u(y,xB1B)=(y/xB1B).
u(xB1B)的数值取自表A5.
4u(y,xB1B)(mgdmP2P)*1000CNAS-GL06:2006第91页共148页2006年06月01日发布2006年07月01日实施例A5:原子吸收光谱法测定陶瓷中溶出的隔详细讨论A5.
1介绍这是评价一个经验方法的不确定度的范例,该经验方法(BS6748)为测定从陶瓷、玻璃、玻璃-陶瓷和搪瓷器皿中溶出的金属.
该方法使用原子吸收光谱仪(AAS)通过用4%(v/v)醋酸水溶液浸泡测定从陶瓷表面溶出的铅或镉的量.
从这个分析方法获得的结果仅与用相同方法得到的其它结果相比较.
A5.
2步骤1:技术规定英国标准BS6748:1986"陶瓷、玻璃、玻璃-陶瓷和搪瓷器皿中溶出的金属限量"给出了完整的测定程序,并形成被测量的技术规定.
下面给出的仅仅是大概描述.
A5.
2.
1仪器和试剂的技术规定影响不确定度的试剂规格:·新配制的4%(v/v)冰醋酸水溶液,用水将40ml冰醋酸稀释至1升.
·4%(v/v)醋酸溶液中铅标准溶液的浓度为1)11000(±mgl.
图A5.
4浸取金属程序·4%(v/v)醋酸溶液中镉标准溶液的浓度为1)5.
0500(±mgl.
实验用玻璃仪器要求至少是B级,并且在测定过程中在4%醋酸溶液中不会溶出可检测到含量的铅或镉.
原子吸收光谱仪要求其检测限为:铅0.
2mglP-1P,镉0.
02mglP-1P.
A5.
2.
2程序图A5.
4图示说明了整个测定程序.
影响不确定度评估的技术规定:ⅰ)样品在(22±2)℃的条件下放置,适当时("类别1"的产品),测量样品的表面积.
如在本例中,表面积是2.
37dm(表A5.
1和A5.
3列出了本例的实验数据).
ⅱ)将(22±2)℃的4%v/v醋酸溶液倒入经预处理的样品中,使溶液填充的高度为距离样品溢出处1mm,可从样品上端边缘处测量,或者距离样品的平端或斜边的最边缘处6mm.
ⅲ)记录使用4%v/v醋酸溶液的量,精确至±2%(本例使用了332ml醋酸).
准备表面处理用4%V/V醋酸填充样品浸泡均匀搅拌浸泡液AAS测定AAS校准结果制备校准标准溶液CNAS-GL06:2006第92页共148页2006年06月01日发布2006年07月01日实施ⅳ)样品在(22±2)℃的条件下放置24小时(测镉时要放置在黑暗中),并采取适当的措施防止挥发损失.
ⅴ)放置后,搅拌溶液使其足够均匀,取一部分测试样,必要时进行稀释(稀释系数为d),选用适当的波长在AA仪器上进行分析,本例中是最小二乘法校准曲线.
ⅵ)计算结果,报告在总浸取液中铅和/或镉的量,对于类别1的产品,用每平方分米表面积含多少毫克铅或镉的方式表示,对于类别2和3的产品,用每升体积含多少毫克铅或镉的方式表示.
注:通过邮寄可从BSI客户服务部获取BS6748:1986完整的复印件,地址:389ChiswickHighRoad,LondonW44ALEngland.
电话:+44(0)2089969001.
A5.
3步骤2:识别和分析不确定度来源步骤1描述了"经验方法".
如果这个方法在指定范围内使用,方法的偏差被定义为零.
因此偏差的评估与实验室的操作有关,而与方法固有的偏差无关.
因为还没有一个有证标准物质用于这个标准方法,偏差的总体控制与影响结果的方法参数的控制有关.
这些影响量是时间、温度、质量和体积等.
稀释后醋酸溶液中铅或镉的浓度CB0B用原子吸收光谱法测定,计算公式如下:1000)(BBAC=1lmg其中:CB0B:在浸取液中铅或镉的浓度[mglP-1P]AB0B:浸取液中金属的吸光度BB0B:校准曲线的截距BB1B:校准曲线的斜率对于本例所考虑的类别1的产品,经验方法要求结果用每单位面积溶出的铅或镉的质量r来表示,r的计算式如下:dBaBAdaVCrVVL==100L0)(V2dmmg其附加参数是:r:每单位面积溶出的铅或镉的质量r[2dmmg]VBLB:浸取液的体积[l]CNAS-GL06:2006第93页共148页2006年06月01日发布2006年07月01日实施aBvB:容器的表面积[]2dmd:样品的稀释系数上述被测量公式的前面部分被用于绘制基本因果图(图A5.
5).
图A5.
5初步因果关系图对于本经验方法,目前尚无有证标准物质用于评估实验室的操作.
因此所有可能的影响量都要考虑,如温度、浸泡时间和酸度.
为了考虑附加的影响量,公式中需加入各自的修正因子,因此扩大为:temptimeacidVLfffdaVcr=0这些修正因子包含在已修正的因果图中(图A5.
6).
图中显示为影响cB0B的因素.
注:该标准所允许的温度范围会引入不确定度,这是由于被测量技术规定不完善而产生.
符合经验方法以及实际可行时,考虑温度的影响允许对被报告的结果范围进行评估.
尤其要注意由不同操作温度引起的结果变化,因为是在按照技术规定要求测试而得到的结果,因此不能合理地认为是偏差.
5.
4步骤3:量化不确定度来源这个步骤的目的是对先前识别的每一个不确定度的来源进行量化.
可以用实验数据或基于良好的假定来进行量化.
U稀释系数d校准曲线填充温度校准读数结果r1长度2长度面积c(0)a(V)dV(L)CNAS-GL06:2006第94页共148页2006年06月01日发布2006年07月01日实施对于本例,无需稀释浸取液,因此不用考虑其对不确定的影响.
U体积UVBLB图A5.
6添加了隐含假设(修正因子)的因果关系图填充体积:经验方法要求容器被溶液填充至"距离边缘1mm以内".
对于典型的饮用和厨房用具,1mm将代表器皿高度的1%.
因此容器被填充的体积为99.
5±0.
5%(即大约是容器体积的0.
995±0.
005).
温度:醋酸的温度必须在22±2℃,由于与容器相比液体具有更大的体积膨胀,这个温度范围导致体积测量的不确定度.
假定温度分布为矩形分布,则332ml体积的标准不确定度是:ml08.
032332101.
24=***读数:记录体积VBLB要求的准确度在2%范围内,实际上使用量筒时允许约1%的不准确性(即0.
01VBLB).
假定是三角形分布来计算标准不确定度.
校准:体积校准是根据制造商的技术规格进行的,500ml量筒有±2.
5ml的偏差,假设为三角形分布,计算标准不确定度.
本例中体积为332ml,四个不确定度分量按下式合成:mlVuL83.
1)65.
2()633201.
0(08.
0)6332005.
0()(2222=+*++*=校准曲线f(酸)f(时间)f(温度)填充温度校准读数结果r1长度2长度面积c(0)a(V)dV(L)CNAS-GL06:2006第95页共148页2006年06月01日发布2006年07月01日实施U镉浓度UcB0使用手工绘制的校准曲线计算溶出镉的量.
用1)5.
0500(±mgl镉标准溶液中配制五个标准溶液,其浓度分别为0.
1、0.
3、0.
5、0.
7和0.
9mglP-1P.
使用线性最小二乘法拟合曲线程序的前提是假定横坐标的量的不确定度远小于纵坐标的量的不确定度,因此通常的cB0B不确定度计算程序仅仅与吸光度不确定度有关,而与校准溶液不确定度无关,也不与从同一溶液中逐次稀释产生必然的相关性.
然而在本例中,校准标准溶液的不确定度足够小以至可以忽略.
五个校准标准溶液分别被测量三次,结果见表A5.
2.
表A5.
2:校准结果浓度(mglP-1P)1230.
10.
0280.
0290.
0290.
30.
0840.
0830.
0810.
50.
1350.
1310.
1330.
70.
1800.
1810.
1830.
90.
2150.
2300.
216表A5.
3:镉溶出量分析的中间值和不确定度描述数值标准不确定度u(x)相对标准不确定度u(x)/xcBoB浸取液中镉的含量0.
26mglP-1P0.
018mglP-1P0.
069VBLB浸取液的体积0.
332l0.
0018l0.
0054aBVB器皿的表面积2.
37dmP2P0.
06dmP2P0.
025fBacidB酸浓度的影响1.
00.
00080.
0008fBtimeB浸泡时间的影响1.
00.
0010.
001fBtempB温度的影响1.
00.
060.
06校准曲线为:ABj:B第BiB个校准标准溶液的第j次吸光度BBCAiij0+=CNAS-GL06:2006第96页共148页2006年06月01日发布2006年07月01日实施Ci:第i个校准标准溶液的浓度BB1B:斜率BB0B:截距值标准偏差BB1B0.
24100.
0050BB0B0.
00870.
0029图A5.
7两次测量的线性最小二乘法拟合和不确定度区间线性最小二乘法拟合曲线的相关系数r为0.
997.
拟合曲线见图A5.
7.
残余标准偏差S是0.
005486.
测量浸出溶液两次,浓度cB0B为0.
26mglP-1P,附录E3详细说明了与线性最小二乘法拟合曲线程序有关的不确定度u(cB0B)计算.
因此这里只是简述不相同的计算步骤.
xxScCnPBScu200)(11)(++=2.
1)5.
026.
0(15121241.
0005486.
02++=10018.
0)(=mglCu残差标准偏差S为:0.
250.
200.
150.
050.
00.
00.
20.
40.
60.
81.
0吸光率鋀的浓度[mg/l]CNAS-GL06:2006第97页共148页2006年06月01日发布2006年07月01日实施005486.
02)]([2101=+=∑=nCBBASjnjj以及2.
1)(1==∑=njjxxcCS其中:BB1B:斜率P:测试cB0B的次数n:测试校准溶液的次数CB0B:浸出液中镉的浓度c:不同校准标准溶液的平均值(n次)i:下标,指第几个校准溶液j:下标,指获得校准曲线的测量次数U面积aUBUVUB长度测量:测量样品容器的尺寸计算其总的表面积为2.
37dmP2P,因为样品近似于圆筒但不规则,在95%置信水平中测量偏差估计在2mm范围内.
典型的尺寸介于1.
0dm和2.
0dm之间,其估计的尺寸测量不确定度为1mm(95%的数值除以1.
96后).
典型的面积测量需要高和宽两个长度尺寸(即1.
45dm和1.
64dm).
面积:由于样品没有完整的几何形状,因此面积计算也有不确定度,在本例中,在95%置信水平时估计有另外5%的分量.
长度测量和面积测量的不确定度分量按通常方式合成.
22296.
137.
205.
001.
001.
0)(*++=vau206.
0)(dmauv=U温度影响UfBtempB已进行了温度对陶瓷器皿溶出金属影响的一些研究P(1-5)P.
一般来说,温度影响是相当大的并且随着温度变化,溶出金属呈指数级上升趋势,直至达到极限值.
只有一个研究P1P给出20-25℃温度范围的影响.
从图形资料上看,在接近25℃的温度附近金属溶出量随温度的变化近于线性,其斜率约为5%℃P-1P.
经验方法允许±2℃的范围导致温度系数fBtempB为1±0.
1.
假定为矩形分布,将其转换为标准不确定CNAS-GL06:2006第98页共148页2006年06月01日发布2006年07月01日实施度:06.
031.
0)(==tempfuU时间影响fUBUtimeU对于相对较慢的过程,如浸泡过程,溶出量将大约与时间的微小变化成正比.
Krinitz和Franco发现在浸泡过程的最后6个小时中浓度的平均变化在86mglP-1P时大约是1.
8mglP-1P,即约占0.
3%/小时,因此对于(24±0.
5)小时的浸泡时间.
CB0B需要用系数UfUBUtimeUB进行修正:1±(0.
5X0.
003)=1±0.
0015.
这是矩形分布,产生的标准不确定度为:001.
030015.
0)(=timefuU酸浓度UfBacidB有一个研究酸浓度对铅溶出影响的结果显示,当浓度从4%改变为5%v/v时,某一特定批陶瓷的铅溶出量从92.
9变为19.
101mgl,acidf变为097.
09.
929.
929.
101=或近似0.
1.
而另一个研究使用热浸泡方法,结果显示类似的变化(浓度从2%变为6%v/v时,铅含量有50%的改变).
假定这个影响近似于与酸浓度成线性,估计酸浓度的每变化1%v/v,fBacidB约有0.
1的变化.
在另一个实验中,使用标准NaOH滴定的滴定法建立了该酸浓度和它的标准不确定度()vvuvv/008.
0/%996.
3=,.
采用该酸浓度的不确定度为0.
008%v/v,则fBacidB的不确定度为0008.
01.
0008.
0=*,因为该酸浓度的不确定度已表示为标准不确定度,这个值可被直接作为与fBacidB有关的不确定度.
注:原则上,不确定度值需要对上述单个研究是足够代表所有陶瓷情况的假定进行修正,当然,目前的数据已给出了对不确定度数量的合理评估.
A5.
5步骤4:计算合成标准不确定度假定没有稀释,则单位面积镉溶出量为:temptimeacidvLfffdaVcr=02dmmg中间值和标准不确定度被收集于表A5.
3,将这些数据代入:2036.
00.
10.
10.
137.
2332.
026.
0=****=mgdmr为了计算相乘表示式(见上)的合成标准不确定度,将标准不确定度的每个分量代入下式:CNAS-GL06:2006第99页共148页2006年06月01日发布2006年07月01日实施22222200)()()()()()()(+++++=temptemptimetimefacidacidvvLLcffuffuffuaauVVuCCurru095.
006.
0001.
00008.
0025.
00054.
0069.
0222222=+++++=20034.
0095.
0)(==mgdmrruc表A5.
4给出了计算合成标准不确定度的更简单的电子表格方法,附录E给出了该方法的详细说明.
图A5.
8图示了不同参数和影响量对测量不确定度的贡献,将每个分量的大小(表5.
4中的C13至H13)与合成不确定度(B16)进行比较.
扩展不确定度UB(r)B通过使用包含因子2计算得到:UBrB=0.
0034X2=0.
007mgdmP-2因此按照BS6748:1986标准测量镉溶出量为:(0.
036±0.
07)mgdmP-2上述不确定度计算使用的包含因子为2.
图A5.
8溶出镉测定的不确定度A5.
6范例5的参考资料1,B.
Krinitz,V.
Franco,J.
AOAC56869-875(1973)2,B.
Krinitz,J.
AOAC61,1124-1129(1978)3,J.
H.
Gould,S.
W.
Butler,K.
W.
Boyer,E.
A.
Stelle,J.
AOAC66,610-619(1983)4,T.
D.
Seht,S.
Sircar,M.
Z.
Hasan,Bull.
Environ.
Cintam.
Toxicol>10,51-56(1973)5,J.
H.
Gould,S.
W.
Butler,E.
A.
Steele,J.
AOAC66,1112-1116(1983)+f(temp)f(time)f(tacid)a(V)V(L)c(O)r12034u(y,xB1B)=(y/xB1B).
u(xB1B)的数值取自表A5.
4u(y,xB1B)(mgdmP2P)*1000CNAS-GL06:2006第100页共148页2006年06月01日发布2006年07月01日实施表A5.
4:镉溶出量分析的不确定度电子表格法计算ABCDEFGH10cLVVaacidftimeftempf2数值0.
260.
3322.
371.
01.
01.
03不确定度0.
0180.
00180.
060.
00080.
0010.
06450c0.
260.
2780.
260.
260.
260.
260.
266LV0.
3320.
3320.
33380.
3320.
3320.
3320.
3327Va2.
372.
372.
372.
432.
372.
372.
378acidf1.
01.
01.
01.
01.
00081.
01.
09timef1.
01.
01.
01.
01.
01.
0011.
010tempf1.
01.
01.
01.
01.
01.
01.
061112r0.
0364220.
0389430.
0366190.
0355230.
0364510.
0364580.
03860713),(ixyu0.
0025210.
000197-0.
0008990.
0000290.
0000360.
0021851422),()(ixyuyu,1.
199E-56.
36E-63.
90E-88.
09E-78.
49E-101.
33E-94.
78E-61516)(ruc0.
0034在第二行从C2到H2输入参数值,它们的标准不确定度在下一行(C3:H3),电子表格将C2:H2值复制到第二列B5:B10处,使用这些数值后的结果(r)值在B12中,C5显示了CB0B的值,等于C2加上其在C3的不确定度,使用C5:C10值计算的结果在C12中给出,D列和H列按照类似的步骤,13行(C13:H13)显示了C12:H12行减去B12值的结果,在14行(C14:H14)是13行(C13:H13)的值的平方,其和的结果在B14中给出,B16是合成标准不确定度,它是B14的平方根.
CNAS-GL06:2006第101页共148页2006年06月01日发布2006年07月01日实施例A6:动物饲料中粗纤维的测定概要目的用法定的标准方法测定粗纤维.
测试程序测试程序是一个标准的程序,图A6.
1列出了一般步骤.
对空白样进行重复测试以获得空白修正.
被测量纤维含量以与样品重量的百分比表示:acbCfibre100)(*=a:样品质量(约1g)b:测试过程中灰化后的质量损失(g)c:空白测试中灰化后的质量损失(g)不确定度来源的识别图A6.
9给出了完整的因果图.
图A6.
1纤维测试不确定度分量的量化实验室的实验表明在实验室内部,实验方法以可以采用协同研究的复现性数据的方式执行.
通常没有其它显著分量.
在低含量时需要考虑所使用的特定干燥程序.
对结果不确定度的典型评估列入表A6.
1内(以标准不确定度表示).
表A6.
1:合成标准不确定度纤维含量(%w/w)标准不确定度u(CBfibreB)(%w/w)相对标准不确定度u(CBfibreB)/CBfibreB2.
531.
0115.
029.
022=+0.
1250.
40.
08100.
60.
06研碎和称量样品加酸消化加碱消化干燥并称量残留灰化并称量残留结果CNAS-GL06:2006第102页共148页2006年06月01日发布2006年07月01日实施例A6:动物饲料中粗纤维的测试详细讨论A6.
1介绍在方法范围中粗纤维被定义为不溶于酸和碱介质的不含脂肪的有机物.
程序是标准化的,其结果可直接使用,改变程序就改变被测量,因此这是经验方法的一个例子.
该法定方法已有协同研究数据(重复性和复现性).
所述的精密度实验已计划作为方法性能内部评估的一部分.
本方法没有合适的标准物(即被相同方法所验证).
A6.
2步骤1:技术规定对于许多分析方法,被测量的技术规定最好是即全面描述分析方法的不同阶段,并提供被测量的公式.
程序从消化、过滤、干燥、灰化和称重的复杂程序,在图A6.
2中均有简要说明.
对空白坩埚的重复试验也使用该程序.
目的是消化大多数的组分,而留下所有不被消化的物质.
有机物被灰化,留下无机残留物,干燥的有机/无机残留物重量与灰化后残留物重量之差就是"纤维含量".
主要步骤如下:ⅰ)研磨样品使其通过1mm筛子;ⅱ)称量1g重样品置于已称重的坩埚中;ⅲ)加入规定浓度和体积的酸消化试剂,煮沸一定时间(按规定),过滤和冲洗残渣;ⅳ)加入标准碱消化试剂,煮沸一定时间,过滤,用丙酮冲洗;ⅴ)在标准规定的温度下干燥至恒重("恒重"在公布的方法中没有定义;也没有给出其它的干燥方法,如空气循环或残留物的分散);ⅵ)记录干燥残渣的重量;ⅶ)在规定温度下灰化至"恒重"(实际上在内部实验研究后决定灰化的时间);ⅷ)称量灰化残渣的重量,减去空白坩堝中残留物重量后计算纤维的含量.
被测量纤维含量以占样品重量的百分比表示:acbCfibre100)(*=a:样品重量(g),称取约1g样品来分析CNAS-GL06:2006第103页共148页2006年06月01日发布2006年07月01日实施b:测试过程中灰化后的重量损失(g)c:空白测试中灰化后的重量损失(g)图A6.
2动物饲料中纤维测定法规方法步骤流程图A6.
3步骤2:识别和分析不确定度来源识别不确定度各种来源,以该方法因果图进行说明(见图A6.
9),这个图按照附录D的程序被简化并去掉重复的部分.
加上去掉不显著的分量,得到A6.
10简化的因果图.
因为本方法有以前协同研究和内部研究的数据,使用这些数据与评估不确定度分量有密切关系,所以下面做进一步的讨论.
A6.
4步骤3:不确定度分量的量化协同试验结果研磨样品使其通过1mm筛子称量坩埚作空白验称1g样品置于坩埚中加助滤剂、防泡沫剂,接着加入150ml沸腾HB2BSOB4B剧烈沸腾30分钟过滤并用3*30ml沸水冲洗加入防泡沫剂,接着加入150ml沸腾KOH剧烈沸腾30分钟过滤并用3*30ml沸水冲洗使用真空装置,用3*25ml丙酮冲洗在130°C干燥至衡重在475~500°C灰化至衡重计算粗纤维含量%CNAS-GL06:2006第104页共148页2006年06月01日发布2006年07月01日实施该方法是协同试验项目,在协同试验研究中分析了五种不同的代表典型的纤维和脂肪含量的饲料.
协同研究的参加者按本方法的全部过程进行了试验,包括样品的研磨.
从协同试验中获得的重复性和复现性估计值见表A6.
2.
作为方法内部评估的一部分,实验计划用与协同试验所分析样品纤维浓度类似的饲料进行重复性(批精密度)评估,结果见表A6.
2,每一个内部重复性评估是基于5次重复实验.
表A6.
2:方法的协合研究和内部重复性检查的结果汇总纤维含量(%w/w)协同试验结果内部重复性标准偏差样品平均值复现性标准偏差重复性标准偏差A2.
30.
2930.
1980.
193B12.
10.
5630.
3580.
312C5.
40.
3900.
2640.
259D3.
40.
3470.
2320.
213E10.
10.
5750.
3910.
327内部得到的重复性估计值与协同试验获得的相当,表明在这个特定实验室中的方法精密度与参加协同试验的实验室的方法精密度相近,因此在评估方法的不确定度中可以使用协同试验的复现性标准偏差.
为了评估不确定度,我们要考虑是否有未被协同试验包括的其它影响因素需要指出来.
协同试验包括不同样品基体和样品前处理,因为参加者得到的样品需要在分析测试前研碎.
与基体影响和样品前处理有关的不确定度不需要另外的考虑,其它影响结果的参数与方法中使用的萃取和干燥条件有关,这些要分别研究以保证实验室偏差得到控制(与复现性标准偏差比较很小).
要考虑的参数在下面讨论.
灰化的质量损失由于本方法没有合适的标准参考物质,通过考虑与方法每一个步骤有关的不确定度来进行内部偏差的评估.
有几个因素对灰化后重量损失有关不确定度有贡献:酸浓度碱浓度CNAS-GL06:2006第105页共148页2006年06月01日发布2006年07月01日实施酸消化时间碱消化时间干燥温度和时间灰化温度和时间试剂浓度和消化时间在以前发表的文章中已研究了酸浓度、碱浓度、酸消化时间和碱消化时间的影响,在这些研究中,评估了该参数变化对分析结果的影响,对于每个参数计算了灵敏系数(最终结果变化与参数变化的比率)和参数的不确定度.
表A6.
3中不确定度比表A6.
2中复现性数值小,例如对于含有2.
3%w/w纤维的样品,其复现性标准偏差为0.
293%w/w,与酸消化时间变化有关的不确定度评估值为0.
021%w/w(2.
3*0.
009),因此我们可以安全地忽略掉与这些方法参数的变化有关的不确定度.
表A6.
3与方法参数有关的不确定度参数灵敏度相关系数P注1P参数的不确定度最终结果的不确定度RSDP注4P酸浓度0.
23(mollP-1P)P-1P10013.
0mollP注2P0.
00030碱浓度0.
21(mollP-1P)P-1P10023.
0mollP注2P0.
00048酸消化时间0.
0031minP-1P2.
89minsP注3P0.
0090碱消化时间0.
0025minP-1P2.
89minsP注3P0.
0072注1:通过制作纤维含量变化与试剂浓度或消化时间的关系图评估灵敏度相关系数.
使用线性回归来计算分析结果随参数变化的变化率.
注2:从制备酸碱溶液所使用的容量瓶的精密度和准确性的估计值以及温度影响等来计算酸和碱溶液浓度的标准不确定度,例子A1-A3中有计算该溶液浓度不确定度的进一步例子.
注3:方法规定消化时间为30分钟,消化时间控制在±5分钟范围内,这是一个矩形分布,除以3后换算为标准不确定度.
注4:以相对标准偏差表示最终结果的不确定度,通过将参数的不确定度乘以其灵敏度相关系数计算得到.
干燥温度和时间没有以往的数据可用.
方法要求样品在130℃温度下干燥至"恒重".
本例中样品在130℃温度下干燥3小时然后称重.
再重复干燥一个小时并称重.
在这个实验室中恒重被定义为连续两次称重的CNAS-GL06:2006第106页共148页2006年06月01日发布2006年07月01日实施变化小于2mg.
在内部实验研究中,四种饲料的平行样在110、130、150℃温度下干燥3和4小时后称重.
在大多数情况下,3小时和4小时之间的样品重量变化小于2mg,因此被作为干燥引起重量变化的不确定度的最坏情况进行评估,±2mg的范围为矩形分布.
除以3被转化为标准不确定度,因此干燥至恒重后所记录重量的不确定度是0.
00115g,方法规定样品重量为1g,对于1g样品,干燥至恒重的不确定度对应于0.
115%w/w纤维含量的标准不确定度.
这个不确定度来源独立于样品中纤维含量,因此不管样品中纤维浓度是多少,对于每一个样品的不确定度评估都将有固定的0.
115%w/w的分量.
对于所有纤维含量,这个不确定度小于复现性标准偏差,除了很低纤维浓度以外,都小于SBRB值的1/3,所以这个不确定度来源通常可以忽略.
对于低纤维浓度,这个不确定度大于SBRB值的1/3,因此在不确定度评估中要给予额外考虑(见表A6.
4).
表A6.
4:合成标准不确定度纤维含量(%w/w)标准不确定度u(CBfibreB)(%w/w)相对标准不确定度uu(CBfibreB)/CBfibreB2.
531.
0115.
029.
022=+0.
1250.
40.
08100.
60.
06灰化温度和时间方法要求样品在475℃至500℃温度下灰化至少30分钟,已经公布的关于灰化条件影响的研究,该研究涉及在450℃30分钟到650℃3小时之间的一系列不同灰化温度/时间条件下纤维含量的测量.
在不同条件下获得的纤维含量没有显著性差异,因此灰化温度和时间的小的变化对最终结果的影响可被忽略掉.
空白灰化后质量损失对于这个参数没有实验数据可用,然而正如上面所讨论的,这个参数变化产生的影响很小.
A6.
5步骤4:计算合成标准不确定度这是有协同试验数据的经验方法的实例.
内部重复性已进行了评估,并且与协同试验所预测的相当,因此可以使用从协同试验得到的sBRB值.
步骤3的讨论可以得出这样的结论,除了低纤维浓度下干燥条件的影响之外,所识别的其它不确定度来源与sBRB值比都很小,在这类情况下,可根据从协同试验获得的复现性标准偏差sBRB值进行不确定度评估.
对于纤维含量为2.
5%w/w的样品,应附加考虑与干燥条件有关的不确定度.
CNAS-GL06:2006第107页共148页2006年06月01日发布2006年07月01日实施标准不确定度表A6.
4给出了一定范围的纤维浓度典型的标准不确定度.
扩展不确定度表A6.
5给出了典型的扩展不确定度,计算时使用包含因子为2,置信水平约为95%.
表A6.
5:扩展不确定度纤维含量(%w/w)扩展不确定度U(CBfibreB)(%w/w)扩展不确定度CV(%)2.
50.
622550.
816101.
212CNAS-GL06:2006第108页共148页2006年06月01日发布2006年07月01日实施粗纤维(%)干燥温度干燥时间空白灰化后重量损失(c)样品重量(a)精密度灰化前坩埚重量天平线性天平校准坩埚重量碱消化P*P酸消化P*P灰化温度灰化时间天平校准天平线性坩埚重量天平校准天平线性样品称量精密度萃取精密度灰化精密度称量精密度灰化后样品和坩埚的重量灰化温度灰化时间天平线性天平校准消化条件萃取时间消解密酸消化酸体积酸浓度干燥温度干燥时间天平线性天平校准消化条件萃取时间沸腾速率碱消化酸体积酸浓度灰化前样品和坩锅的重量灰化后重量损失(b)*加到"酸消化"和碱消化"上的分支为清楚起见省略掉了,因为影响它的因素与影响样品的因素相同(即消化条件、酸浓度等).
图A6.
9动物饲料中纤维测定的因果关系图灰化后坩埚重量坩埚重量坩埚重量CNAS-GL06:2006第109页共148页2006年06月01日发布2006年07月01日实施样品称量精密度萃取灰化精密度称量精密度粗纤维(%)干燥温度干燥时间空白灰化后重量损失(c)精密度灰化前坩埚重量碱消化P*P酸消化P*P灰化温度灰化时间灰化后样品和坩埚的重量灰化温度灰化时间消化条件萃取时间沸腾速率酸消化酸体积酸浓度干燥温度干燥时间消化条件萃取时间凝胶速率碱消化酸体积酸浓度灰化前样品和坩锅的重量灰化后重量损失(b)*加到"酸消化"和碱消化"上的分支为清楚起见被省略掉了,因为影响它的因素与影响样品的因素相同(即消化条件、酸浓度等).
图A6.
10简化的因果关系图灰化后坩埚重量CNAS-GL06:2006第110页共148页2006年06月01日发布2006年07月01日实施例子A7使用双同位素稀释和电感耦合等离子体质谱测定水中的铅含量A7.
1介绍本例说明了在使用同位素稀释质谱(IDMS)和电感耦合等离子体质谱(ICP-MS)技术时,如何应用不确定度测定水样中铅的含量.
双IDMS概述:IDMS是被Comitèconsultatifpourlaquantetèdematière(CCQM)认可的一种可能成为基准的测量方法.
因此它有如何计算被测量的详细表达式.
作为最简单的例子,用具有富集同位素参考物质的标定示踪物进行同位素稀释,测量示踪样、样品和已知样品量与示踪样的混合物b中的同位素率.
下式给出了样品中元素的含量xc:∑∑=iyyixxxxbbbbyyxyyxiiiiRKRKRKRKRKRKmmcc)()(1111(1)其中:xc和yc分别是样品和示踪样的元素含量(用符号c代替k代表含量是为了避免与K-因子和包含k因子同符号而引起的混乱).
xm和ym是样品和示踪样的质量,yxRR,和bR是同位素含量比率.
下标x,y和b分别代表样品、示踪样和混合物.
选择一种通常在样品中最丰富的一种同位素,所有其它同位素的含量表示为它的相对比率.
选择一对特定的同位素(参考同位素和在示踪样中含量最丰富的同位素),作为监测比率,如)(/)(206208pbnpbn.
ixR和iyR分别是样品和示踪样中所有可能的同位素含量比率.
对于参考同位素,这个比率是1.
iiyxKK,和bK是对样品、示踪样和混合物中具体同位素含量比率的质量甄别校正因子.
使用标定的同位素标准物质,根据公式(2)测定K因子.
biasKKK+=0;其中observedcertifiedRRK=0(2)其中0K是时间为0时的质量甄别校正因子,在测量时,当K因子用于校正不同时间测量的校正比率时,biasK作为偏差因子就开始生效.
biasK还包括其它的可能的偏差,如乘数死时间校正,基体效应等.
certifiedR是同位素标准物质证书上的标定的同位素含量比率,observedR是该同位素标准物质观测值.
CNAS-GL06:2006第111页共148页2006年06月01日发布2006年07月01日实施在IDMS实验中,使用电感耦合等离子体质谱(ICP-MS),质量碎片随时间改变,因此要求对公式(1)中所有的同位素含量比率分别作质量甄别校正.
富含一种特定同位素的标定标准物质常常是难以获得的.
为了克服这个问题,经常使用"双"IDMS.
程序是使用一个没有很好确定特性,但是富含同位素示踪样的物质与一个具有天然同位素组成的标定标准物质(标识为z)联合使用.
此标定的、天然组成的物质作为基准定量分析标准品.
使用两种混合物,混合物b是样品和富含同位素示踪样的混合物,如公式(1)所示.
为了进行"双"IDMS,第二个混合物b′是由含量为1zc的基准分析标准品与富集物质为y制备而成.
这里给出了与公式(1)相近的表达式:∑∑=iyyizzzzbbbbyyzyyziiiiRKRKRKRKRKRKmmcc).
().
(.
.
.
.
1111'''''(3)其中:zc是基准分析标准溶液的元素含量,zm是制备新的混合物时的基准分析标准的质量,'ym是富含同位素示踪样溶液的质量.
1,,''zbbKRK和1zR分别是新的混合物和基准分析标准物的K因子和同位素比率.
下标z表示该基准分析标准.
将公式(1)除以公式(3)得:∑∑∑∑=iyyizzzzbbbbyyzyyiyyixxxxbbbbyyxyyzxiiiiiiiiRKRKRKRKRKRKmmcRKRKRKRKRKRKmmccc)()()()(11111`111'''''(4)化简这个公式,引入分析程序中的空白cBblackB,得到:blankizzixxbbyyzzbbxxbbbbyyyzxyzxcRKRKRKRKRKRKRKRKRKRKmmmmcciiii*=∑∑)()('''''11111111(5)这是最终的一个公式,在其中yc已经被消掉.
在本测量中,同位素含量的比率R的下标数字代表了以下实际同位素含量比率:)(/)()(/)(20620732062081pbnpbnRpbnpbnR==)(/)()(/)(20620442062062pbnpbnRpbnpbnR==作为参考,所有参数汇总在表A7.
1中.
CNAS-GL06:2006第112页共148页2006年06月01日发布2006年07月01日实施A7.
2步骤1:技术规定测量的总的步骤见表A7.
2中.
所涉及的计算和测量在下面说明.
表A7.
1IDMS参数概述参数描述参数描述mBxB混合物b中样品的质量(g)mByB混合物中富集标准样品的质量(g)mP'PByB混合物b'中富含同位素标准样品的质量(g)mBzB混合物b'中基准分析标准的质量(g)cBxB样品x的含量(molgP-1P或molgP-1P)P注1PcBzB基准分析标准z的含量(molgP-1P或molgP-1P)P注1PcByB标准样品y的含量(molgP-1P或molgP-1P)P注1PcBblankB程序空白中所观察的含量(molgP-1P或molgP-1P)P注1PRBbB所测得的混合物b的比率,n(P208PPb)/n(P206PPb)KBbBRBbB的质量偏差校正因子RP'PBbB所测得的混合物b'的比率,n(P208PPb)/n(P206PPb)KP'PBbBR'BbB的质量偏差校正因子RBy1B所测得的富含同位素的标准样品中富含同位素与标准同位素的比率KBy1BRBy1B的质量偏差校正因子RBziB基准分析标准中的所有比率,RBz1,BRBz2B等KBziBRBziB的质量偏差校正因子RBxiB样品中所有的比率KBxiBRBxiB的质量偏差校正因子RBx1B所测得的样品x中富集同位素与标准同位素的比率RBz1B同RBxi,B但是在基准分析标准中注1:含量的单位通常在文件中说明.
表A7.
2总程序步骤描述1制备基准分析标准2制备混合物b'和b3测定同位素比率4计算样品中Pb的含量cBxB5评估cBxB的不确定度含量xc的计算程序在这个测定水中铅的过程中,对每个混合物b'(定量分析标准+标准样品)和b(样品+标准样品)CNAS-GL06:2006第113页共148页2006年06月01日发布2006年07月01日实施各制备了四份混合样.
这就给出了4个xc值.
每一次测量按表A7.
2步骤1至4所述的进行.
报告值cBXB是4次重复测试的平均值.
计算摩尔质量由于某些元素(如Pb)的同位素组成的天然变化,将会对含量zc产生影响,所以必须测定基准分析标准的摩尔质量M.
注意若zc以1lgmo为单位那就没有影响.
对于一个元素E,其摩尔质量M(E)在数值上等于元素E的原子量)(EAr,原子量可以根据通式进行计算:∑∑===piipiiirREMREA11)()((6)其中iR是元素E所有的真实的同位素含量比率.
)(EMi是列表的核素质量.
注意公式(6)中的同位素含量比率必须是绝对的比率,也就是,它必须用质量甄别进行修正.
使用合适的下标得到公式(7).
在进行计算时,核素质量)(EMi取自文献数值2,而比率izR和0K-因子)(ziKo要进行测定(见表A7.
8),得到:11121034.
207.
)(.
.
)1,(====∑∑gmolRKEMRKAssaypbMpizzpiizzziiii(7)测定K-因子和同位素含量比率为了修正质量甄别,使用了公式(2)中所规定的修正因子K.
可使用有已标定同位素组成的标准物质计算0K-因子.
此种情况中,用同位素已标定的标准物质NISTSRM981监测0K-因子可能的变化.
0K-因子在修正比率前后进行测定.
一个典型的样品次序是:1.
(空白),2.
(NISTSRM981),3.
(空白),4.
(混合物1),5.
(空白),6.
(NISTSRM981),7.
(空白),8.
(样品)等.
空白测定不仅仅是用来做空白修正,也可以用来监测空白计数.
只有当空白样品计数率稳定并回到正常水平才能做新的测试.
注意在测试前应把样品、混合物、示踪样品和定量分析标准品稀释至适合的含量水平.
比率测定的结果、所计算的0K-因子和biasK的结果汇总在表A7.
8中.
基准分析标准的制备和含量zc的计算CNAS-GL06:2006第114页共148页2006年06月01日发布2006年07月01日实施制备两个基准分析标准,其中铅是从化学纯度为W=99.
999%的不同金属铅块上获得的.
两块铅块都来自同一批的高纯度铅.
将两个铅块样品溶于3/3:110wHNOwml:水中后微微加热,然后进一步稀释.
两个混合物各由这两个定量分析标准来制备.
下面介绍一个分析标准的一些数值.
将0.
36544g(1m)铅用).
05.
0(13llmHNO水溶液溶解并稀释至总重为14.
1961=dg.
此溶液定为标准1(Assay1).
取标准1质量为0292.
12=mg,用).
05.
0(13llmHNO水溶液稀释到总重为931.
992=d克,这个溶液定为标准2(Assay2).
标准2的Pb含量zc根据公式(8)计算如下:1181122lg092605.
0lg102605.
9)1Assay,(1=*==momopbMdwmdmczμ(8)混合物的制备示踪样品的质量分数是已知的,约为20μgPb/g的溶液.
也已知该样品中Pb的质量分数在这个范围内.
表A7.
3列出了本例中使用的两个混合物的称量数值.
表A7.
3混合物bb'所使用的溶液加料样品样品加料样品样品参数mBy1BmBxBmBy2BmBzB质量(g)1.
13601.
04401.
06541.
1029程序空白Blankc的测量在本例中用外标法测定程序空白,一个更复杂的方法是将富含同位素的示踪样品加入到空白中,并用与样品同样的方法处理它.
在本例中,只使用高纯度的试剂,这样会产生混合物的极端比率并随之产生富含同位素示踪程序的低可靠性.
外标校准程序空白被测定4次,得到Blankc值为17lg105.
4*moμ,按A类不确定度评估,其标准不确定度为17lg100.
4*moμ.
计算未知含量xc将测定和计算的数据(表A7.
8)代入公式(5)中得到1.
053738.
0=gmolcxμ.
四次重复测定值见表A7.
4.
A7.
3步骤2和3:不确定度来源的识别和量化CNAS-GL06:2006第115页共148页2006年06月01日发布2006年07月01日实施不确定度计算的思路假如把公式(2)、(7)和(8)代入最终的IDMS公式(5),参数数目之多使得公式无法处理.
为了使公式简单化,将0K-因子和标准定量分析溶液的含量和它们相关的不确定度分别处理,然后才代入IDMS公式(5).
在本例中,这样将不影响xc最终的总不确定度.
出于实际的考虑,公式做这样的简化是可行的.
为了计算总的标准不确定度)(xcuc,使用其中一次测量的数值(如在A7.
2中所述).
将使用附录E所描述的电子表格方法计算xc的总不确定度.
表A7.
4)lg(1moCxμ重复试验1(本例子)0.
053738重复试验20.
053621重复试验30.
053610重复试验40.
053822平均值0.
05370实验标准偏差(S)0.
0001K-因子的不确定度:i)0K的不确定度K用公式(2)计算得到,作为一个例子使用1xK的数值得出KB0B:9992.
01699.
21681.
2)(10===observedcertifiedRRxK(9)为了计算0K的不确定度,我们首先查看标准证书,证书给出的比率是2.
1681,在95%置信度时其不确定度为0.
0008.
为了把基于95%置信度的不确定度转化为标准不确定度,我们把以上的不确定度除以2得到标准不确定度0004.
0)(=certifiedRu.
观察到的数量比率,)(/)(206208pbnpbnRobserved=的标准不确定度为0.
0025(rsd).
K-因子的总不确定度计算如下:002507.
0)0025.
0()1681.
20004.
0()())((22110=+=xKxKuc(10)CNAS-GL06:2006第116页共148页2006年06月01日发布2006年07月01日实施这明显看出标准证书的比率对不确定的贡献是可以忽略的.
因此,所测量的比率observedR的不确定度将用作0K的不确定度.
biasK的不确定度:引入偏差因子(biasK)是因为考虑到质量甄别因子的数值可能的偏离.
正如在上述因果图和公式(2)中所见到的,每一个K-因子都有偏差.
这些偏差的数值在本例中是不知道的,所以取0.
当然每一个偏差均有不确定度,当计算最终不确定度时必须考虑这些偏差不确定度.
原则上,象公式(11)中使用的偏差一样,通过引用公式(5)以及参数1yK和1yR可以表示这个原理:KKLK+=1).
()((110ybiasxRyKyKc.
……(11)所有偏差值),,(zixiyiKbias在(0±0.
0001)之间.
该估计值是基于铅IDMS测试的长期经验,所有的),,(zixiyiKbias参数没有详尽地包括在表7.
5,表7.
8或公式5中,但是它们在所有不确定度计算中都被使用.
表A7.
5数值标准不确定度类型P注1PKBbiasB(zi)00.
001BRBZ1B2.
14290.
0054AKB0B(z1)0.
99890.
0025AKB0B(z3)0.
99930.
0035AKB0B(z4)1.
00020.
0060ARBz2B10ARBz3B0.
91470.
0032ARBz4B0.
058700.
00035AMB1B207.
9766360.
000003BMB2B205.
9744490.
000003BMB3B206.
9758800.
000003BMB4B203.
9730280.
000003BCNAS-GL06:2006第117页共148页2006年06月01日发布2006年07月01日实施注1:A类(统计评估)或B类(其它)所称质量的不确定度在本例情况下,称量是由一个专业的质量计量实验室进行的.
称量使用的程序是一种运用所校正的砝码和比较仪的高低射标技术.
该技术对每一个样品质量测量至少重复6次.
应用了浮力校正.
在本例中没有运用化学计算法和杂质修正.
称量证书上的不确定度作为标准不确定度,见表A7.
8.
标准分析溶液含量zc的不确定度:i)Pb原子量的不确定度:首先计算标准溶液[标准1(Assay1)]的摩尔质量的总不确定度,表A7.
5中的数值已知或已被测得.
根据公式(7),摩尔质量的计算如下:4433221144433322111)1,(zzzzzzzzzzzzzzzRKRKRKRKMRKMRKMRMRKAssayPbM++++++=(12)为了计算标准溶液中Pb摩尔质量的总标准不确定度,使用附录E中的电子表格模型.
每个比率和0K测量八次:使用附录E电子表格模型计算出的不确定度为10010.
0gmol,得出摩尔质量12103.
207)1,(=gmolAssayPbM,.
ii)测定zc中总标准不确定度的计算为了计算标准溶液中Pb含量zc的不确定度,使用表A7.
2的数据和公式(8).
称量不确定度从其证书中得到,见表A7.
3.
公式(8)中使用的所有参数及其不确定度见表A7.
6.
使用公式(8)计算zc含量.
根据附录D.
5,zc的总标准不确定度由计算得到:000028.
0)(=zccu.
其标准不确定度为1lg000028.
0moμ(用rsd表示为0.
03%)时,得到1lg092606.
0=moczμ.
使用附录E的电子表格模型,计算重复测试1的)(xccu.
重复测试1的不确定度评估将作为该测量的不确定度.
由于公式(5)的参数多,没有用电子表格显示出来.
参数的数值和其不确定度以及xc的总不确定度见表A7.
8.
表A7.
6数值不确定度铅块质量,mB1B(g)0.
365440.
00005CNAS-GL06:2006第118页共148页2006年06月01日发布2006年07月01日实施第一次稀释液的总质量dB1B(g)196.
140.
03取第一次稀释液质量mB2B(g)1.
02920.
0002第二次稀释液的总质量dB2B(g)99.
9310.
01金属铅块的纯度w(质量分数)0.
999990.
000005标准物质中Pb的摩尔质量M(gmolP-1P)207.
21040.
0010A7.
4步骤4:计算总标准不确定度表A7.
7显示四次重复测定的平均值和实验标准偏差.
数值取自表A7.
4和表A7.
8.
在IDMS和很多非日常分析中,测试程序的完整统计控制需要耗费无数的资源和时间.
检查一些不确定度来源是否已被忽略的好的方法是将A类评估得出的不确定度与四次重复测试的实验标准偏差相比较.
假如实验标准偏差大于按A类评估的不确定度分量,表明测定过程没有完全被理解.
作为一种近似,使用表7.
8的数据,通过取总的实验不确定度的92.
2%,计算A类评估的实验不确定度的总和,为1lg00041.
0moμ.
这个数值明显高于1lg00010.
0moμ的实验标准偏差(见表A7.
7).
这表明实验的标准偏差已被按A类评估的不确定度分量所包括,因此无需进一步考虑来自混合物制备的按A类评估的不确定度分量.
然而可能有与混合物制备有关的偏差.
在本例中,混合物制备中可能的偏差与主要不确定度来源相比时,可判定混合物制备的偏差是不显著的.
水样中Pb的含量为:1lg)00036.
005370.
0(±=mocxμ该结果可提供使用包含2因子时的扩展不确定度.
表A7.
7重复测定1重复测定1-4的平均值cBxB=0.
05374cBxB=0.
05370molgP-1Pu(cBxB)=0.
00018s=0.
00010P注1PmolgP-1P注1:此为实验标准不确定度,不是平均值的标准偏差.
例子A7参考资料1.
T,Cvita,Metrologia,1996,33,35-392.
G,AudiandA.
H.
Wapstra,Nuclearphysics,A565(1993)CNAS-GL06:2006第119页共148页2006年06月01日发布2006年07月01日实施表A7.
8参数不确定评估数值实验不确定度(注1)对总不确定度的贡献uBcB(%)最终不确定度(注2)对总不确定度的贡献uBcB(%)ΣKBbiasBB00.
001P注3P7.
20.
001P注3P37.
6cBzBB0.
0926050.
0000280.
20.
0000280.
8KB0B(b)A0.
99870.
002514.
40.
000889.
5KB0B(b')A0.
99830.
002518.
30.
0008811.
9KB0B(x1)A0.
99920.
00254.
30.
000882.
8KB0B(x3)A1.
00040.
003510.
00120.
6KB0B(x4)A1.
0010.
00600.
00210KB0B(y1)A0.
99990.
002500.
000880KB0B(z1)A0.
99890.
00256.
60.
000884.
3KB0B(z3)A0.
99930.
003510.
00120.
6KB0B(z4)A1.
00020.
00600.
00210mBxBB1.
04400.
00020.
10.
00020.
3mBy1BB1.
13600.
00020.
10.
00020.
3mBy2BB1.
06540.
00020.
10.
00020.
3mBzBB1.
10290.
00020.
10.
00020.
3RBbBA0.
293600.
0007314.
20.
00026P注4P9.
5R'BbBA0.
50500.
001319.
30.
0004612.
7RBx1BA2.
14020.
00544.
40.
00192.
9RBx2BCons.
100RBx3BA0.
91420.
003210.
00110.
6RBx4BA0.
059010.
0003500.
000120RBy1BA0.
000640.
0000400.
0000140RBz1BA2.
14290.
00546.
70.
00194.
4RBz2BCons.
100RBz3BA0.
91470.
003210.
00110.
6RBz4BA0.
058700.
0003500.
000120cBblankBA4.
5*10P-7P4.
0*10P-7P02.
0*10P-7P0cBxB0.
053740.
000410.
00018ΣABcontrib.
.
B=92.
2ΣBBcontrib.
.
B=7.
8ΣABcontrib.
.
B=60.
4ΣBBcontrib.
.
B=39.
6注1:实验不确定度在计算时没有考虑每个参数的测量次数.
注2:在最终不确定度的计算中考虑了测定次数.
在本例中所有按A类评估的参数测定了8次.
其标准不确定度要除以8.
注3:该数值是一个KBbiasB的数值.
用参数ΣKBbiasB来代替所有的KBbiasB(zi,xi,yi)的列表,这些数值都是(0±0.
001).
注4:每个混合物其RBbB测定8次,总共测定32次.
当没有混合物之间的变异时,如本例,在此模型中所有混合物均进行四次重复测定时,就能得出了所有32个测定值.
这要花费很多时间,在本例中因为它不会显著地影响不确定度,这就没有做.
CNAS-GL06:2006第120页共148页2006年06月01日发布2006年07月01日实施附录B定义总则B.
1测量准确度测量结果与被测量的真值之间的一致程度[H.
4].
注1:"准确度"是一个定性概念.
注2:不要用"精密度"代替"准确度".
B.
2精密度在规定条件所获得的独立测量结果之间的一致程度[H.
5].
注1:精密度只取决于随机误差的分布,而与真值或规定值无关.
注2:精密度的度量通常用不精密度术语表示,并计成测量结果的标准偏差.
大的标准偏差反映了小的精密度.
注3:"独立测量结果"意味着所获得的测量结果不受以前任何同样或类似物体的测量结果所影响.
定量测量精密度关键取决于规定的条件.
重复性和重现性条件就是一组规定的极端条件.
B.
3真值与给定的特定量的定义一致的值[H.
4].
注1:这是一个通过完善的测量才能获得的值.
注2:真值按其本性是不确定的.
注3:"真值"要与不定冠词"a"而不是定冠词"the"一起使用,因为与给定的特定量的定义一致的值可能有很多.
B.
4约定真值对于给定目的具有适当不确定度,赋予特定量的值,有时该值是约定采用的[H.
4].
例:a)在给定地点,由参考标准复现而赋予该量的值可作为约定真值.
b)常数委员会(CODATA)1986年推荐的阿伏加德罗常数值注1:"约定真值"有时称为指定值、最佳估计值、约定值或参考值.
注2:常用某量的多次测量结果来确定约定真值.
B.
5影响量不是被测量但对测量结果有影响的量[H.
4].
例1:用来测量长度的千分尺的温度;mol1100221367.
623*CNAS-GL06:2006第121页共148页2006年06月01日发布2006年07月01日实施2:交流电位差幅值测量中的频率;3:测量人体血液样品血红蛋白浓度时的胆红素的浓度.
测量B.
6被测量作为测量对象的特定量[H.
4].
注:对被测量的详细描述,可要求包括对其他有关量(如时间、温度和压力)作出说明.
B.
7测量以确定量值为目的的一组操作[H.
4].
B.
8测量程序进行特定测量时所用的,根据给定的测量方法具体叙述的一组操作[H.
4].
注:测量程序(有时被称为测量方法)通常记录文件中,并且足够详细,以使操作者在进行测量时不再需要补充资料.
B.
9测量方法进行测量时所用的,按类别叙述的一组操作逻辑次序[H.
4].
注:测量方法可按不同方式分类,如替代法、微差法、零位法.
B.
10测量结果由测量所得到的赋予被测量的值.
注:当给出"测量结果"时,应说明它是示值、未修正测量结果或已修正测量结果,还应表明它是否为几个值的平均值.
注2:在测量结果的完整表述中应包括测量不确定度的信息.
不确定度B.
11(测量)不确定度表征合理地赋予被测量之值的分散性,与测量结果相联系的参数.
注1:此参数可以是诸如标准偏差(或其指定倍数),或说明了置信水平的区间半宽度.
注2:测量不确定度由多个分量组成.
其中一些分量可用测量列结果的统计分布估算,并用实验标准偏差表征.
另一些分量则可用基于经验或其他信息的假定概率分布估算,也可用标准偏差表征.
注3:测量结果应理解为被测量之值的最佳估计,而所有的不确定度分量均对分散性有贡献,包括那些由系统效应引起的分量,如与修正值和参考标准有关的分量.
B.
12溯源性CNAS-GL06:2006第122页共148页2006年06月01日发布2006年07月01日实施通过一条具有规定不确定度的不间断的比较链,使测量结果或测量标准的值能够与规定的参考标准,通常是与国家测量标准或国际测量标准联系起来的特性[H.
4].
B.
13标准不确定度)(ixu:以标准偏差表示的测量结果ix的不确度[H.
2].
B.
14合成标准不确定度)(yuc:当测量结果y是由若干个其他量的值求得时,按其他各量的方差或协方差算得的标准不确定度[H.
2].
B.
15扩展不确定度确定测量结果区间的量,合理赋予被测量之值分布的大部分可望含于此区间[H.
2].
注1:该部分可认为是该区间包含概率或区间的置信水平.
注2:为了将特定的置信水平与扩展不确定度确定的区间联系起来,需要对表征测量结果及其合成不确定度的概率分布作出清晰或不容置疑的假设.
表征该区间的置信水平只能达到该假设所允许的合理程度.
注3:扩展不确定度U按下述公式由合成标准不确定度cu和包含因子k计算得到:cukU*=B.
16包含因子为求得扩展不确定度,对合成标准不确定度所乘之数字因子[H.
2].
注:包含因子一般在2-3范围内.
B.
17(不确定度的)A类评定用对观测列进行统计分析的方法,来评定标准不确定度[H.
2].
B.
18(不确定度的)B类评定用不同于观测列进行统计分析的方法,来评定标准不确定度[H.
2].
误差B.
19(测量)误差测量结果减去被测量的真值[H.
4].
注:由于真值不能确定,实际上用的是约定真值.
B.
20随机误差测量的结果与在重复性条件下,对同一被测量进行无限多次测量所得结果的平均值之差[H.
4].
CNAS-GL06:2006第123页共148页2006年06月01日发布2006年07月01日实施注1:随机误差等于误差减去系统误差.
注2:因为测量只进行有限次数,故可能确定的只是随机误差的估计值.
B.
21系统误差在重复性条件下,对同一被测量进行无限多次测量所得结果的平均值与被测量的真值之差[H.
4].
注1:系统误差等于误差减去随机误差.
注2:如真值一样,系统误差及其原因是不能完全获知.
统计术语B.
22算术平均值x:一个样品n个结果的算术平均值.
nxxnii∑==,1B.
23样品标准偏差s:由一个样品的n个结果得出的总体标准偏差σ的估计值.
1)(12=∑=nxxsniiB.
24平均值的标准偏差从总体中抽取n个数值,其平均值的标准偏差由下式给出:nssx=术语"标准误差"和"平均值的标准误差"也曾用来表达同样量.
B.
25相对标准偏差(RSD)RSD:由一个样品的n个结果得出总体标准偏差的估计值除以该样品的平均值,也称变异系数.
通常也用百分比表示:xsRSD=CNAS-GL06:2006第124页共148页2006年06月01日发布2006年07月01日实施附录C:分析过程中的不确定度C.
1为了识别分析过程中的不确定度可能来源,将分析过程分解成一组通用的步骤是有帮助的:1:抽样2:样品制备3:有证标准物质对测试系统的影响4:仪器的校准5:分析(数据采集)6:数据处理7:结果的表达8:结果的解释C.
2这些步骤可按对不确定度的贡献进一步分组.
下面所列出的内容,虽然不一定全面,但提供应考虑因素的指南.
1.
抽样-均匀性-具体的抽样策略的影响(例如,随机抽样、分层随机抽样、比例抽样等)-媒介移动的影响(尤其是密度选择)-媒介的物理状态(固体、液体、气体)-温度和压力影响-抽样过程是否影响组成例如,在抽样系统中的差色吸附.
2.
样品制备-均匀性和/或二级抽样的影响-干燥-碾磨-溶解-萃取-污染-衍生(化学影响)-稀释误差CNAS-GL06:2006第125页共148页2006年06月01日发布2006年07月01日实施-(预)浓缩-物种形成影响的控制3.
有证标准物质对测量系统的影响-有证标准物质的不确定度-有证标准物质是否与样品匹配4.
仪器的校准-使用有证标准物质的仪器校准误差-标准物质及其不确定度-校准用的物质是否与样品匹配-仪器的精密度5.
分析-自动分析仪的进位-操作者的影响,例如色盲、视差、其他系统误差-基体、试剂或其他被分析物的干扰-试剂的纯度-仪器参数的设置,例如积分参数-重复性实验的精密度6.
数据处理-平均-修约的控制-统计-运算法则(模型拟合,例如线性最小二乘法)7.
结果的表达-最终结果-不确定度的估计-置信水平8.
结果解释-对照限值/范围-法规的符合性-目的的适用性CNAS-GL06:2006第126页共148页2006年06月01日发布2006年07月01日实施附录D:分析不确定度来源D.
1介绍通常有必要将分析方法有关的所有不确定来源分析出来并加以记录.
将这一过程系统化通常是有用的,既可保证考虑范围的全面性,又可避免重复过高.
下面的步骤(基于以前出版的方法[H.
14]),提供了一种合适的、系统地分析不确定度产生原因的可能方法.
D.
2方法的原理D.
2.
1该策略分成两步:·识别对结果的影响因素实际上,通过使用因果图(有时称作Ishikawa或"鱼骨"图)[H.
15]来进行必要的系统分析.
·简化并解决重复的情况首次列出的内容要进行精简并且保证影响因素没有不必要地重复列出.
D.
3因果分析D.
3.
1构造因果图的原理在其他地方加以详述.
所使用的步骤如下:1.
写出结果的完整公式.
该公式中的参数构成因果图的主要分支.
几乎有必要增加一个对总偏差(通常以回收率来表示)修正的主要分支.
适当时,推荐在此本步骤中增加此分支.
2.
考虑方法的每一步骤,并且从主要影响因素之外来考虑,在因果图上进一步增加其他因素,如环境及基体的影响.
3.
对每一个分支,增加有贡献的影响因素直至影响因素变得足够小,即直到对结果的影响可忽略.
4.
解决重复问题,并重新安置,澄清影响因素,将有关的有不确定度来源编成组.
在该步骤在单独的精密度分支上集合所有精密度内容是便利的.
D.
3.
2因果分析的最后步骤要求进一步说明.
对每个输入参数的贡献量进行详细分析时,自然会产生重复性问题.
例如,对任何影响因素,重复性实验的变异性总是存在的,至少在名义上.
这些影响因素总体上会对方法的总体方差有贡献.
因此,假如已有这样考虑了,就不需单独列出.
同样,通常用同一台仪器称量物质,会导致校准不确定度的重复计算.
出于这些考虑,就有了下述精简因果图的附加规则(虽然它完全等同地适用于任何系统地列出的影响因素).
·取消影响因素:两者均要去掉.
例如,在差减称量中,称量两次,两次均受天平"零偏差"的影响,"零偏差"将由于重量差而消除.
因此,可在分别列出的称量有关分支中取消.
·类似的影响因素,同样时间:合成一个单一输入量.
例如:许多输入量的重复性变化能合成一CNAS-GL06:2006第127页共148页2006年06月01日发布2006年07月01日实施个总的重复性精密度"分支".
尤其需要注意,每一次测量单独操作间的变异性可以合成,而对整批次操作间的变异性(例如仪器校准)只有用批次间精密度度量时才能观测到.
·不同的情况:重新标注.
通常会发现类似命名的影响因素实际上是指类似测量的不同情况.
在进行下一步之前,必须清楚区分.
D.
3.
3这种类型的分析不会导致单一结构的列表.
在目前的例子中,温度既可视为所测密度的直接影响因素,也可视为是对此比重瓶中的物质所测质量的影响因素,两者均可成为首次结构内容.
实际上这不影响方法的使用性.
假如所有重要的影响因素在列表的某个地方只出现过一次,总的一套方法仍然有效.
D.
3.
4一旦因果图分析完成,一种适当的做法是回到结果的原始公式,并增加任何新的项(例如温度)到公式中.
D.
4例子D.
4.
1本步骤通过参照简化了的直接密度测量例子来说明.
考虑直接测量乙醇密度d(EtOH)的例子,通过称量合适的带刻度容器的皮重mtare以及加了乙醇后的毛重mgross来获得已知体积乙醇的质量.
密度按下式计算d(EtOH)=(mgross-mtare)/V为了清晰,仅考虑三个影响因素:仪器校准、温度和每次测量的精密度.
图D1-D3用图表的方式说明了这过程.
D.
4.
2因果图由一个结果的分支结构组成,其最终只导致一个结果.
对目前的目的而言,该结果就是具体的分析结果(图D1的'd(EtOH)').
指向该结果的'各分支'是贡献因素,包括具体的中间测量结果和其他因素,诸如环境或基体影响.
每一个分支接着又有自己的贡献因素.
这些"因素"包含影响结果的各种因素,无论是变量或常数.
这些因素的不确定度都明显地对结果的不确定度有贡献.
D.
4.
3图D1显示了应用步骤1-3直接获得的一种可能的图表.
主要分支是公式中的参数,对各参数的影响因素由次分支来表示.
注意,有两个"温度"影响因素,3个精密度影响因素和3个"校准"影响因素.
D.
4.
4图D2显示了按照第二条规则(相同影响因素/时间)将精密度和温度各自组合在一起.
温度可作为影响密度的单一因素,而每次测量的变异性均贡献给整个方法的重复实验所观测到的变异性.
D.
4.
5按照第一个精简规则(取消),两个称量的校准偏差相互抵消了,可以去除(图D3).
D.
4.
6最后,余下的校准分支需要分成两个(不同)分量,一个可能是由于天平响应的非线性,另一个是与体积测量有关的校准不确定度.
线性=inearity精密度CNAS-GL06:2006第128页共148页2006年06月01日发布2006年07月01日实施图D1:首次列表温度d(EtOH)精密度m(毛)m(皮)线性偏差校准温度线性偏差校准校准体积精密度图D2:类似影响因素组合d(EtOH)温度m(毛)温度精密度线性偏差校准精密度体积校准精密度温度线性偏差校准m(皮)精密度图D3:取消d(EtOH)温度m(毛)线性偏差校准体积校准精密度线性偏差校准m(皮)偏差取消同一天平:CNAS-GL06:2006第129页共148页2006年06月01日发布2006年07月01日实施附录E:有用的统计程序E.
1分布函数下列表格显示了如何从两个最重要的分布函数的参数来计算标准不确定度,并给出它们能被使用的环境.
例:一个化学家估计一个影响因素不小于7或不大于10,并感到具体数值位于这个区间的任何地方,但不知道是否区间的任何部分比另一部分更加可能.
这是描述了区间2a=3(半宽a=1.
5)的矩形分布函数的情况.
使用下面矩形分布的函数,标准不确定度的估计值可计算出来.
使用上面的区间,a=1.
5,标准不确定度的结果87.
0)35.
1(=.
矩形分布图形在下述情况使用不确定度·证书或其他技术规定给出了界限,但无规定置信水平(例如:25ml±0.
05ml)·估计值是以最大区间(±a)形式给出的,但没给出分布的形状3)(axu=三角形分布图形在下述情况使用不确定度·所获得的有关x的信息不仅限于矩形分布.
靠近x的数值比接近两边界的更加可能.
·估计值是以最大区间(±a)形式作出并具有对称分布6)(axu=1/2ax2a(=±a)1/ax2a(=±a)CNAS-GL06:2006第130页共148页2006年06月01日发布2006年07月01日实施正态分布图形在下述情况使用不确定度估计值是对随机变化过程的重复测量作出的不确定度是以标准偏差s,相对标准偏差xs/或方差系数CV%给出,未给出分布不确定度以95%(或其他)置信水平,区间为x±c给出,未规定分布(95%置信水平)(99.
7%置信水平)E.
2电子表格方法计算不确定度E.
2.
1电子表格软件可用来简化第8节的计算.
该程序利用微分法的近似数字方法,并且只要求知道用来导出最终结果(包括任何必要的校正因子或影响)的计算以及参数的数值和它们的不确定度.
此处所描述的是按照Kragten[H.
12]的方法.
E.
2.
2在不确定度))xx,xu(y(n21K的表达式中假如y(xB1B,xB2B…xBnB)对ix是线性或与ix相比)(ixu是小的,偏导)/(xyi可近似为:乘以)(ixu获得因ix不确定度引起的y的不确定度),(ixyu,得:sxu=)(xCVxuxssxusxu===100%)()/()()(2/)(cxu=3/)(cxu=∑∑=+=nkikikixxuxyxynixiuxiy,1,2),(,1)()()())((xuxyxuxyxyiiiii+≈),,()))((,(),(2121xxxxyxxuxxxyxyuniniiiLLLL+≈1/ax2σCNAS-GL06:2006第131页共148页2006年06月01日发布2006年07月01日实施因此,),(ixyu只是分别用)]([iixux+和ix计算出来的y之差.
E.
2.
3线性或小的iixxu/)(的假设并不是在所有的情况下都充分得到满足.
尽管如此,当对评估)(ixu的值进行必要的近似进行考虑时,该方法确实提供对于实际用途的可接受的准确性.
参考文献H.
12对这点讨论更详细并且建议核查假设有效性的方法.
E.
2.
4基础的电子表格设立如下,假设结果y是四个参数p、q、r和s的函数.
ⅰ)在电子表格A栏内输入p、q等值以及计算y的公式.
将栏A中y的各变量复制到其他各栏(见图E2.
1).
如图所示将不确定度u(p)、u(q)等的值放在第一行是方便的.
ⅱ)将u(p)加到单元B3的p中,将u(q)加到单元C4的q中等等,见图E2.
2.
重新计算电子表格后,单元B8就变成),),((Lrqpupf+(在图E2.
2和E2.
3中用),,,'(Lrqpf表示),单元C8就变成),),(,(Lrquqpf+等.
ⅲ)在第九行输入第8行减A8(例如,单元B9变成B8-A8),给出u(y,p)的值为u(y,p)=f(p+u(p),q,r…)-f(p,q,r…)等ⅳ)为了得到y的标准不确定度,各个分量分别平方,并加在一起,然后开平方根,即通过在10行输入u(y,p)2(图E2.
3),并且将这些和的平方根放在A10.
即,单元A10相当于下列公式SQRT(SUM(B10+C10+D10+E10))它给出了y的标准不确定度.
E2.
5单元B10、C10等的内容显示了y不确定度的各个不确定度分量的平方分量u(y,xi)2=2))((iixuc,因此容易看出哪一个分量是显著的.
E2.
6随着个别参数值改变或不确定度的更新,可直接进行即时计算.
在上面的步骤i),不是直接将栏A复制到栏B至E中,而是通过引用而将p至s值复制,即单元3B至3E均引用3A、单元4B至4E引用4A等.
图2.
1的水平箭头表明第3行引用情况.
注意单元8B至8E还分别引用列B至列E的值,如图2.
1的B列竖箭头所示.
在上面的步骤ii)中,通过引用加上第一行的引用值(如图E2.
1箭头所示).
例如单元3B变成,13BA+单元4C变成14CA+等.
对参数或不确定度的改变将立刻在8A的总结果中及10A的合成标准不确定度中反映出来.
E2.
7假如变量是相关的,要在10A的SUM中增加必要的附加项.
例如,假如p和q相关,其相关系CNAS-GL06:2006第132页共148页2006年06月01日发布2006年07月01日实施数为),(qpr,则附加项),(),(),(2qyupyuqpr***要在开平方根之前加到所计算的和中.
因此通过电子表格中增加适当的附加项就可以很容易将相关性包括进去.
图E2.
1ABCDE1u(p)u(q)u(r)u(s)23ppppp4qqqqq5rrrrr6sssss78y=f(p,q…)y=f(p,q…)y=f(p,q…)y=f(p,q…)y=f(p,q…)91011图E2.
2ABCDE1u(p)u(q)u(r)u(s)23pp+u(p)ppp4qqq+u(q)qq5rrrr+u(r)r6sssss+u(s)78y=f(p,q…)y=f(p′…)y=f(…q′…)y=f(…r′…)y=f(…s′…)9u(y,p)u(y,q)u(y,r)u(y,r)1011CNAS-GL06:2006第133页共148页2006年06月01日发布2006年07月01日实施E.
3线性最小二乘法校准的不确定度E.
3.
1分析方法或仪器通常是通过观察被分析物x的不同浓度的响应值y来校准的.
在大多数情况下,这种关系被认为是线性的,即xbbyo1+=1.
3.
EEq利用该校准线,可通过样品中被分析物产生的响应值yBobsB,由下式测得其浓度xBpredB:1/)(bbyxoobspred=2.
3.
EEq通常通过对一组n对数值),(iiyx的加权或未加权最小二乘法回归来确定常数1b和obE.
3.
2为了获得估计值predx的不确定度,四种主要不确定度来源需要考虑.
·测量y时的随机变化,既影响标准响应值iy,又影响被测量的响应值obsy·导致标准值赋值ix误差的随机效应·ix和iy值可能受恒定的未知偏移的影响,例如当x值取自常备溶液的连续稀释时所产生的偏移·线性的假设未毕有效上面因素中,在正常操作中最显著的是y的随机变化,该种来源的不确定度评估方法将在此详述.
其它来源也简要地加以考虑以便指出所用的方法.
E.
3.
3由于y的(随机)变化性,预估值xpred的不确定度可按几种方法来评估:从计算所得的方差和协方差来获取.
假如1b和ob的值,它们的方差var(1b),var(ob)以及它们的协方差covar(obb,1)是由最小二乘法图E2.
3ABCDE1u(p)u(q)u(r)u(s)23pp+u(p)ppp4qqq+u(q)qq5rrrr+ur6sssss+u(s)78y=f(p,q…)y=f(p′…)y=f(…q′…)y=f(…r′…)y=f(…s′…)9u(y,p)u(y,q)u(y,r)u(y,s)10u(y)u(y,p)P2Pu(y,q)P2Pu(y,r)P2Pu(y,s)P2P11CNAS-GL06:2006第134页共148页2006年06月01日发布2006年07月01日实施获得,x的方差var(x)通过使用第八章提供的公式,并对该标准公式求微分,得:21112)var(),(cov.
.
2)var(.
)var()var(bbbbarxbxyxoopredpredobspred+++=Eq.
E3.
3以及对应的不确定度)var(),(predpredxyxu=从校准数据获得上述)var(predx的公式可用测量校准函数所使用的一组n个数据点),(iiyx来表示:++=∑∑∑∑)/)()(.
()(1.
/)var()var(22221221iiiiiprediobspredwxwxwxxwbSbyxEq.
E3.
4其中)(,)2()(22fiifiiiyynyywS=∑是第i个点的余差,n是校准的数据点的数目,1b是计算所得的最佳拟合斜率,iw是赋与iy的权以及)~(xxpred是predx与n个21,xx…值的平均值x之间的差.
对未加权数据以及)var(obsy是基于p次测量,公式E3.
4变成:++=∑∑)/)()(()(11.
)var(2212212nxxxxnpbSxipredpredEq.
E3.
5这是例子5所使用的公式:().
)(/)(222∑∑∑==xxnxxSiiixx由用来导出校准曲线的软件提供的信息某些软件给出S的值,不同叫法如RMS误差或残余标准误差.
然后可用在E3.
4或E3.
5中.
然而,一些软件也可从某些新的x值的拟合线上计算所得的y值的标准偏差)(cys,因此这可用来计算)var(predx,对于p=1()()∑∑++=nxxxxnysiipredc/)()(11)(222对照E3.
5,给出[]21/)()var(bysxcpred=Eq.
E3.
6E.
3.
4标准值ix各有自己的不确定度,并通过传播律传给最终结果.
实际上,这些数值的不确定度与系统的响应值iy的不确定度相比通常是小的,因此可以忽略.
由于具体的标准值的不确定度引起的预CNAS-GL06:2006第135页共148页2006年06月01日发布2006年07月01日实施估值predx的不确定度近似值为:nxuxxuiipred/)(),(≈EqE3.
7其中n是用于校准的ix值的数目.
这个表达式可用来检查),(ipredxxu的重要性.
E.
3.
5由于假设y和x的线性关系引起的不确定度通常不会大到要求做额外的评估.
假如余差没有表明与这种假设关系有重大系统偏差,由这种假设所引起的不确定度可忽略(除了导致y方差最终增加的假设外).
假如余差表明有系统偏离趋势,在校准函数有必要包括较高阶项式.
在这些情况下的var(x)的计算方法见标准文本.
基于系统偏离的大小也可做出判断.
E.
3.
6x和y的值可受恒定的未知偏移影响(例如当x的值取自给出不确定度的有证常备溶液的连续稀释时所引起的),假如这些影响对y和x产生的标准不确定度分别为u(y,const)和u(x,const),内插值predx的不确定度为)var()/),((),()(2122xbconstyuconstxuxupred++=Eq.
E3.
8E.
3.
7E.
3.
2所述的四个不确定度分量可用Eq.
E3.
3至Eq.
E3.
8的公式进行计算.
线性校准计算引起的总不确定度可按常规的方式合成这四个分量来计算.
E.
4与被分析物浓度有关的不确定度的表示E4.
1.
引言E4.
1.
1在化学测试中,经常观察到在被分析物浓度的大范围中,在总的不确定度中占支配作用的分量几乎与被分析物的浓度成比例变化,即)(xux∝.
在这类例子中,通常以相对标准偏差或如变化系数(%CV)来引述不确定度是合理的.
E4.
1.
2当不确定度不受浓度的影响,例如在低浓度或被分析物的浓度范围较窄时,引用不确定度的绝对值更合理.
E4.
1.
3在某些情况下,恒定的影响和按比例变化的影响都是重要.
当不确定度随被分析物浓度的变化是重要的以及简单作为一个变化系数报告是不合适时,本节给出了记录不确定度信息的通用方法.
E4.
2方法的基础E4.
2.
1既要考虑到不确定度的随被分析物浓度变化的比例性,又要考虑不随被分析物浓度变化的恒定数值的可能性,使用下列通用公式:CNAS-GL06:2006第136页共148页2006年06月01日发布2006年07月01日实施212).
()(sxsxuo+=[1]其中u(x)是结果x的合成标准不确定度(即,用标准偏差表示的不确定度)sB0B代表对总不确定度的贡献恒定的分量sB1B是比例常数该表达式是基于常用的将两个不确定度分量合成总不确定的方法,并假设分量(os)是常数,而分量)(1xs与结果成比例,图E4.
1展示了该表达式的形式.
注:上述方法只有当可能计算大量的数值时才可行.
当进行实验研究时,通常不能建立相关的抛物线关系.
在这种情况下,通过对在不同的被分析物浓度获取的四个或更多的合成不确定度进行简单的线性回归可获得合适的近似值.
该程序与根据ISO5725:1994进行的重现性和重复性的程序是一致.
有关的表达式为111.
)(sxsxuo+≈.
E4.
2.
2该图可分为大致的区域(见图A至C)A:不确定度受项0s的支配,因此基本恒定,约等于0sB:两项均相当重要.
最终的不确定度比0s或1xs高出许多,可见一部分曲线.
C项1xs占支配地位.
不确定度随x的增加而几乎呈线性增长,因此约等于1xs.
E4.
2.
3注意在许多实际例子中,没有明显的完整的曲线形式.
极常见的是,方法范围所允许的被分析物浓度的整个报告范围落在一个单一图区.
下面更详细介绍的一些特殊例子的结果就是这样.
E4.
3与浓度相关的不确定度数据的表示E4.
3.
1一般来说,不确定度可用0s和1s的每一个值的形式来表示.
在整个方法的范围内可用这些数值提供不确定度的估计值.
当在计算机系统上对描述完整的方法进行计算时,其中应用的通用公式独立于参数的值(其中之一可为0,见下面),这尤其重要.
因此建议,除下面给出的特定情况,或当相关性较强并且不是线性*之外,不确定度用以常量0s表示的数值和以变量1s表示的数值来表示.
*一个非线性相关的的重要例子是仪器噪音对接近仪器性能的上限高吸光度的吸光度测量的影响.
当吸光度是通过透射比率(如在红外光谱中)来计算时尤其明显.
在这种情况下,基线噪音使得高吸光度数值有非常大的不确定度,并且不确定度值增长地比简单的线性估计所预测地更快.
通常的做法是降低吸光度,尤其是通过稀释,使得吸光度的数值刚好落在工作范围内.
此处所使用的线性模型因此变得合适了.
其他例子包括一些免疫测定方法的"s形"响应.
CNAS-GL06:2006第137页共148页2006年06月01日发布2006年07月01日实施E4.
4特殊情况E4.
4.
1不确定度与被分析物的浓度无关(os起支配作用)不确定度通常会有效地独立于所观测的被分析物的浓度,当:·结果接近于零(例如在方法规定的检测限内).
图E4.
1的区域A.
·结果的可能范围(在方法的范围中规定的或在不确定度评估的范围声明中规定的)与所观测到的浓度相比是小的.
在这些情况下,1s的值可记为零.
0s通常是所计算的标准不确定度.
E4.
4.
2不确定度完全与被分析物的浓度相关(1s占支配作用)当结果远大于零(例如,高于"测量限")以及有明显的证据表明在方法范围所允许的被分析物浓度范围内,不确定度随着被分析物的浓度而按比例变化,1xs项起支配作用(见图E4.
1的区域C).
在这种情况下,以及方法范围不包括约为零的被分析物的浓度,0s可合理地记为零,1s是以相对标准偏差表示的不确定度.
E4.
4.
3中间相关在中间的情况,以及尤其当情况对应于图E4.
1区域B时,可采取两种方法:a)使用变量相关更常用的方法是测量、记录和使用0s和1s.
当需要时,不确定度的评估基于所报告的结果.
可行时推荐该方法.
注:见E4.
2注b)使用固定的近似在通常测试中可使用的另一个方法,当:·相关性不强(即,比例性的证据不强),或·所期望的结果范围是中等时.
上面任一情况均导致变化不超过平均不确定度估计值的15%的不确定度,因此基于所期望的结果平均值计算和引用一个不确定度的固定值作为通用值是合理的.
或者用x的平均值或特征值来计算固定的不确定度估计值,并且以此来代替个别计算的估计值,或基于研究覆盖所允许的被分析物浓度的全范围(在不确定度估计值的范围内)的物质研究,得到单一的标准偏差,并且几乎没有证据说明比例性的假设是合理的.
一般应该将它作为零相关的情况,有关的标准偏差记为s.
CNAS-GL06:2006第138页共148页2006年06月01日发布2006年07月01日实施E4.
5测定0s和1sE4.
5.
1在一个项式占支配作用的特殊情况下,分别以标准偏差或相对标准偏差表示的不确定度作为0s和1s的值通常是足够的.
然而,当相关性不甚明显时,有必要在不同的被分析物浓度间接地从一系列不确定度估计值来确定0s和1s.
E4.
5.
2假定从不同的分量进行合成不确定度的计算,其某些分量与浓度有关而另外一些则不相关,通常可通过模拟来研究总不确定度与被分析物浓度的相关性.
程序如下:1.
计算(或通过实验获取)涵盖所允许的全范围内至少十个被分析物浓度的不确定度;2.
画2)(ixu对2ix的图;3.
通过线性回归,获得cmxxu+=22)(的线中m和c的估计值;4.
从cs=0,ms=1计算0s和1s;5.
记录0s和1s.
E4.
6报告E4.
6.
1此处所列出的方法允许评估任何单次结果的标准不确定度.
原则上,当报告不确定度信息时,将以下述形式,[结果]±[不确定度]其中以标准偏差表示的不确定度按上述计算,并且假如必要时扩展(通常乘以因子2)以增加置信水平.
然而,当许多结果一起报告时,给出适合于所报告的所有结果的不确定度估计值是可能的并且完全可接受.
E4.
6.
2表E4.
1给出一些例子.
一系列不同被分析物的不确定度值可按同样的原则有效地计算.
注:当一个"检测限"或"报告限"用来给出以"CNAS-GL06:2006第139页共148页2006年06月01日发布2006年07月01日实施表E4.
1几个例子的不确定度汇总场合占支配的项报告的例子所有结果的不确定度基本恒定0s或固定的近似值(E4.
4.
1或E4.
4.
3a节)标准偏差:扩展不确定度;95%置信水平的区间不确定度通常与浓度成比例1xs(见E4.
4.
2节)相对标准偏差:方差系数(%CV)比例性和不确定度低限值的混合中间例子(节E4.
4.
3)引用%cv或rsd以及标准偏差表示的低限sB0x.
sB1Bu(x)B1.
81.
61.
41.
210.
80.
60.
40.
20012345678不确定度约等于80不确定度比sB0B或x.
s.
都要大得多不确定度约等于x.
sABC结果x图E4.
1不确定度的观察结果的变化CNAS-GL06:2006第140页共148页2006年06月01日发布2006年07月01日实施附录F检测限/测量限的测量不确定度F.
1引言F.
1.
1在低浓度,日益增加的各种影响变得重要,包括,例如·噪音或不稳定基线的存在·干扰物对(毛)信号的干扰作用·所使用的任何分析空白的影响以及·在萃取、分离或净化过程中的损失因为这些影响,随着被分析物浓度降低,与结果有关的相对不确定度趋于增加,首先达到结果的很大一部分,最终达到其(对称的)不确定度区间包含零.
该区域是典型地与给定方法的实际检测限有关.
注:与测量和报告低浓度被分析物有关的术语和约定已经广泛在其他地方加以讨论(有关例子和定义见文献H.
16,H.
17,H.
18).
这里,术语"检测限"只表示检测成为疑问的一种浓度,并且不与任何特定的定义相关联.
F1.
2普遍认为,"检测限"最重要的用途是显示方法性能不足以提供可接受的定量,因此需要改进.
因此,理想时,定量测量不要在这个区域进行.
但是如此多的材料在这个低浓度是重要的,以至不可避免要在这个区域进行测量以及结果的报告.
F1.
3当结果是小的并且与结果相比不确定度是大的时,测量不确定度的ISO指南[H.
2]并没有给出不确定度评估的清晰指引.
确实,本导则第8章所描述的"不确定度传播律"的基本形式在这个区域可能不能精确使用.
该计算基于的假设之一是与被测量的值相比不确定度是小的.
从ISO指南给出的不确定度定义将产生另一个理论上的困难.
尽管在这个区域负的观察值是相当可能,并且甚至相当常见,当被测量是浓度时,所指的含零以下值的分散不能是".
.
.
.
.
.
合理赋予被测量的值",因为浓度本身不能为负.
F1.
4这些困难并未阻止本指南所描述的方法的使用.
但是在解释和报告该区域的测量不确定度结果时要谨慎.
本附录的目的是提供有限的指南补充从其他途径所获得的指南.
注:类似的考虑可适用于其他区域.
例如,摩尔或质量分数接近100%可导致类似的困难.
F.
2观测值和估计值F.
2.
1测量科学的基本原理是结果是真值的估计值.
例如,分析结果开始是以所观测到的信号为单位的,如mV,吸光度单位等.
为传递给更多的读者,尤其是实验室的客户或其他官方,原始数据需要转换成CNAS-GL06:2006第141页共148页2006年06月01日发布2006年07月01日实施化学量,如浓度或物质的量.
这种转换典型地要求校准程序(可包括,如对观测值的修正以及清楚描述的损失的修正).
然而,无论何种转换,所产生的数值仍为观测值或信号,假如实验是正确进行,该观测值数值仍为被测量的最佳估计值.
F2.
2观测值通常不被适用于实际浓度的同样基本限值所限制.
例如,报告一个所观测到的浓度,即低于零的估计值,是完全合理的.
谈到在同样区域内可能观察值的分散也是同样合理.
例如,当对一个没有被分析物存在的样品进行无偏见的测量时,人们应该看到约一半观察值低于零.
换句话说,类似的报告:观测到的浓度=1184.
2±mg观测到的浓度=1192.
4±mg不仅可能,它们应被视为有效的表达.
F2.
3本指南所描述的不确定度评估方法完全适合于观测值的不确定评估.
当报告观测值及其不确定度给一个内行的读者时,即使结果表明一个不可能的物理环境,对最佳值及其不确定度的报告的理解不会有阻碍和矛盾.
确实,在某些情况下(例如,当报告一个被用来修正其他结果的分析空白值时),报告该观测值及其不确定度(不管多大)是绝对必要的.
F2.
4这是真实的情况,无论结果的最终使用是否值得怀疑.
因为只有该观测值和它的不确定度能被直接使用(例如,在进一步计算、在趋势分析或重新解释中),未经审查的观测值应总是可获得.
F2.
5因此理想的情况是报告有效观测值及其不确定度,而不管其值如何.
F.
3经解释的结果和符合性声明F3.
1尽管前面所述的理由,为了最终用户的利益,必须承认许多包含一些解释的分析报告及符合性声明.
典型地,这类解释包括材料中合理存在的被分析物浓度有关的结论.
这类解释是关于真实情况的结论,因此被认为(被最终用户)是符合真正的限值.
因此,其任何不确定的估计值也是"真"值.
F3.
2当最终用途已被充分理解时,以及最终用户因实际上不能了解测量观测值的性质时,其它地方提供的有关报告低浓度结果的一般导则(例如文献H.
16,H.
17,H.
18)可合理地适用.
F3.
3然而进一步的谨慎是贴切的.
许多关于检测能力的文献依赖于重复性观测值的统计.
本指南的读者应该清楚所观察的变化只是罕有作为结果总不确定度的良好指南.
正如处理任何其他领域的结果一样,因此在报告的数值之前应仔细考虑影响该结果的所有不确定度.
CNAS-GL06:2006第142页共148页2006年06月01日发布2006年07月01日实施附录G不确定度的常见来源和数值下表概括了一些不确定度分量的典型例子.
该表给出·具体的被测量或实验程序(测定质量、体积等)·每一例中不确定度的主要分量和来源·测定每一个来源的不确定度所建议的方法·典型的例子该表旨在概括例子,并指出在分析中评估测量不确定度的一般方法.
它们并不旨在全面,所给的值也不可未经独立判断而直接使用.
然而,这些数值可帮助决定某个具体分量是否显著.
CNAS-GL06:2006第143页共148页2006年06月01日发布2006年07月01日实施典型值测量不确定度分量原因测量方法例子数值天平校准不确定度校准的有限准确度将校准证书上声明的值转换为标准偏差4位天平0.
5mg线性i)在有证砝码范围内实验ii)制造商的规格约0.
5*最后一位有效数字日偏移不同,包括温度长期核查称量的标准偏差.
必要时计算成RSD约0.
5*最后一位有效数字可读性显示器或刻度的有限分辨率来自最后一位有效数字0.
5*最后有效位/3重复性变化不同连续样品或核查称量的标准偏差约0.
5*最后一位有效数字密度影响(通常情况下)P注1P校准块/样品密度不配引起空气浮力效应的不同从已知或假设的密度和典型的空气条件来计算钠、镍、铝、有机固体、水、烃1ppm20ppm50-100ppm65ppm90ppm质量密度影响(在真空中)见上计算空气浮力影响并从校准块减去浮力影响100g水10g镍+0.
1g(效应)<1mg(效应)注1:对于基本常数或SI单位定义,通过称量测定质量通常修正到真空中的重量.
在大多数其他实际场合中,引用的重量是基于OIML所定义的常规情况[H.
18].
这个常规是引用在空气密度为32.
1kgm和样品密度为38000kgm的重量,相当于在正常大气条件下处于海平面称量钢.
当样品密度为38000kgm或空气密度为32.
1kgm时,对常规质量的浮力修正是零.
因为空气密度通常非常接近后者,对常规质量的修正通常可忽略.
上表所给出的在常规条件下称量的与密度有关的影响所产生的标准不确定度数值,对于在海平面常规情况下称量,且未考虑修正浮力的初步估计是足够的.
然而,在常规情况下测得的质量与"真实质量"(在真空中)相差0.
1%或更多(见上表最后一行的影响因素).
CNAS-GL06:2006第144页共148页2006年06月01日发布2006年07月01日实施典型值测量不确定度原因测量方法例子值校准不确定度校准有限的准确度制造商规格所声明的转换为标准偏差对ASTM体积为V的A类玻璃仪器,限值约为200/6.
0V10ml(A类)=ml01.
03/02.
0温度与校准温度不同引起的与在标准温度下的体积的差别给出相对标准偏差,其中ΔT是可能的温度范围,α是液体的体积膨胀系数,对于水,α约为14102*k;对于有机液体为13101*k100ml水在规定的操作温度3℃内进行测试为0.
03ml体积(液体)重复性变化不同连续核查排出的体积的标准偏差(通过称量决定)25ml移液管重复充满/称量s=0.
0092ml*假设矩形分布)32/(αΔTCNAS-GL06:2006第145页共148页2006年06月01日发布2006年07月01日实施典型值测量不确定度分量原因测量方法例子数值纯度不纯降低了标准物质的实际数量,活性不纯可干扰测量制造商证书所给出.
标准证书通常给出无法定量的限值.
因此可按矩形分布处理,除以3.
注:当不纯的性质未给出时,可需要额外的考虑或核查来建立干扰限标准邻苯二甲酸氢钾的证书值为99.
9±0.
1%%06.
03/1.
0=浓度(证书)证书给出的标准物质浓度不确定度制造商证书所给出.
标准溶液证书通常给出无法定量的限值;因此应按矩形分布处理,除以3在4%醋酸中的醋酸镉,证书值为1)21000(±mgl12.
132=mgl(RSD为)0012.
0标准物质浓度浓度(从有证材料配制)标准值和中间步骤的不确定度的合成合成前面步骤的数值作为整个过程的RSD三次稀释后醋酸镉:从11000mgl到15.
0mgl0034.
00017.
00021.
00017.
00012.
02222=+++作为RSD*假设为矩形分布CNAS-GL06:2006第146页共148页2006年06月01日发布2006年07月01日实施测量不确定度分量原因测量方法例子数值仪器校准注:该分量是指相对于标准吸光度的吸光度读数,而不是以吸光度的读数对浓度校准校准的有限准确度校准证书给出的作为限值,然后转换为标准偏差吸光度重复性化不同重复测量的标准偏差或QA性能7个吸光度读数的平均,s=1.
6362.
07/63.
1=抽样均匀性不均匀材料的二级抽样通常不能正确的代表整个物质.
注:随机抽样通常导致零偏差.
有必要检查抽样确实是随机的.
i)不同的二级抽样结果的标准偏差(假如与分析准确度比非均匀性较大)ii)从已知或假设的总体参数所估计的标准偏差从假设的2项不均匀性的面包中抽样(见例A4)从72份污染和360份未污染整体中抽样15份:RSD=0.
58CNAS-GL06:2006第147页共148页2006年06月01日发布2006年07月01日实施典型值测量不确定度分量原因测量方法例子值平均回收率萃取很少是完全的,并且可加入或包括干扰物回收率从相当的标准物质或代表性示踪的百分比回收率来计算.
从回收率实验的平均值的标准偏差可得不确定度注:回收率也可直接从以前测量所得的分配系数计算而得面包农药的回收率;42次实验,平均值为90%,s=28%(见例A4)%4342/28=(RSD为0.
048)萃取回收率回收率的重复性变化不同重复实验的标准偏差从成对平行试验的数据所得的面包农药的回收率(见例A4)RSD0.
31`CNAS-GL06:2006第148页共148页2006年06月01日发布2006年07月01日实施附录H参考文献H.
1.
ISO/IEC17025:1999.
GeneralRequirementsfortheCompetenceofCalibrationandTestingLaboratories.
ISO,Geneva(1999).
H.
2.
GuideToTheExpressionOfUncertaintyInMeasurement.
ISO,Geneva(1993).
H.
3.
EURACHEM,QuantifyingUncertaintyinAnalyticalMeasurement.
LaboratoryoftheGovernmentChemist,London(1995).
ISBN0-948926-08-2H.
4.
InternationalVocabularyofbasicandgeneraltermsinMetrology.
ISO,Geneva,(1993).
(ISBN92-67-10175-1)H.
5.
ISO3534:1993.
Statistics–VocabularyandSymbols.
ISO,Geneva,Switzerland(1993).
H.
6.
AnalyticalMethodsCommittee,Analyst(London).
12029-34(1995).
H.
7.
EURACHEM,TheFitnessforPurposeofAnalyticalMethods.
(1998)(ISBN0-948926-12-0)H.
8.
ISO/IECGuide33:1989.
UsersofCertifiedReferenceMaterials.
ISO,Geneva(1989).
H.
9.
InternationalUnionofPureandAppliedChemistry.
PureAppl.
Chem.
,67,331-343,(1995).
H.
10.
ISO5725:1994(Parts1-4and6).
Accuracy(truenessandprecision)ofmeasurementmethodsandresults.
ISO,Geneva(1994).
SeealsoISO5725-5:1998foralternativemethodsofestimatingprecision.
H.
11.
I.
J.
Good,"DegreeofBelief",inEncyclopaediaofStatisticalSciences,Vol.
2,Wiley,NewYork(1982).
H.
12.
J.
Kragten,Analyst,119,2161-2166(1994).
H.
13.
BritishStandardBS6748:1986.
Limitsofmetalreleasefromceramicware,glassware,glassceramicwareandvitreousenamelware.
H.
14.
S.
L.
R.
Ellison,V.
J.
Barwick.
Accred.
Qual.
Assur.
3101-105(1998).
H.
15.
ISO9004-4:1993,TotalQualityManagement.
Part2.
Guidelinesforqualityimprovement.
ISO,Geneva(1993)H.
16.
H.
Kaiser,Anal.
Chem.
4224A(1970).
H.
17.
L.
A.
Currie,Anal.
Chem.
40583(1968).
H.
18.
IUPAC,LimitofDetection,Spectrochim,Acta33B242(1978).
H.
19.
OIMLRecommendationsIR33.
Conventionalvalueoftheresultofweighinginair.
本文件是CNAS实验室的指南性文件,只对化学检测实验室在实施认可准则时提供指引,并不增加对CNAS—CL01:2006《实验室能力认可准则》的要求.
文件编号为CNAS—GL06:2006.
在本文件的翻译和编制得到了深圳出入境检验检疫局、天津出入境检验检疫局和中国电子技术标准化研究所的大力协助,在此表示感谢.
CNAS-GL06:2006第2页共148页2006年06月01日发布2006年07月01日实施目录引言…41.
目的与范围…62.
不确定度…72.
1不确定度的定义…72.
2不确定度的来源…72.
3不确定度的分量…72.
4误差和不确定度…83.
分析测量和不确定度…93.
1方法确认…103.
2方法性能的实验研究…113.
3溯源性…124.
测量不确定度的评估过程…145.
第一步被测量的技术规定…156.
第二步识别不确定度来源…177.
第三步量化不确定度…197.
1引言…207.
2不确定度的评估程序…207.
3以前研究的相关性…217.
4量化单个分量来评估不确定度…217.
5极匹配的有证标准物质…227.
6使用以前的协同方法开发和确认研究数据来评估不确定度……………………227.
7使用实验室内开发和确认研究进行不确定度评估…237.
8经验方法的不确定度评估…267.
9特别方法的不确定度评估…267.
10单个分量的量化…277.
11单个不确定度分量的试验估计…277.
12基于其他结果或数据的评估…287.
13根据理论原理建立模型…297.
14基于判断的评估…297.
15偏差的显著性…31CNAS-GL06:2006第3页共148页2006年06月01日发布2006年07月01日实施8.
第四步计算合成不确定度…318.
1标准不确定度…318.
2合成标准不确定度…328.
3扩展不确定度…359.
不确定度的报告…369.
1总则…369.
2所需要的信息…369.
3报告标准不确定度…379.
4报告扩展不确定度…379.
5结果的数值表示…389.
6与限值的符合性…38附录A例子…40介绍…40例子A1:校准标准溶液的制备…42例子A2:氢氧化钠溶液的标定…50例子A3:酸碱滴定…62例子A4:实验室内部确认研究的不确定度评估面包中有机磷农药的测定75例子A5:原子吸收光谱法测定陶瓷中镉溶出量…88例子A6:动物饲料中粗纤维的测定…100例子A7:使用同位素稀释和电感耦合等离子体质谱测定水中的铅含量……………109附录B定义…119附录C分析过程中的不确定度…123附录D分析不确定度来源…125附录E有用的统计程序…128附录F检测限/测量限的测量不确定度…139附录G不确定度的常见来源和数值…141附录H参考文献…147CNAS-GL06:2006第4页共148页2006年06月01日发布2006年07月01日实施引言很多重要的决策都是建立在化学定量分析的结果基础上,例如,化学定量分析的结果可以用于估计收益、判定某些材料是否符合特定规范或法定限量、或估计货币价值.
当我们使用分析结果来作为决策依据的时候,很重要的一点是必须对这些结果的质量有所了解,换句话说,就是必须知道用于所需目的时,这些结果在多大程度上是可靠的.
化学分析结果的用户,特别是涉及国际贸易领域时,正在受到越来越大的压力减少取得化学分析结果的重复劳动.
达到这个目的的前提是必须建立对由非用户自身机构所得数据的信心.
在化学分析的某些领域,现在已经有一个正式的(经常是法定的)要求,就是要求实验室引进质量保证措施来确保其能够并且正在提供所需质量的数据.
这些质量保证措施包括:使用经确认的分析方法、使用规定的内部质量控制程序、参加水平测试项目、通过根据ISO17025[H.
1]进行的实验室认可和建立测量结果的溯源性.
在分析化学中,过去曾经把重点放在通过特定方法获得的结果的精密度,而不是他们对所定义的标准或SI单位的溯源性.
这种思路导致使用"官方方法"来满足法定要求和贸易要求.
但是,因为现在正式要求建立结果的可信度,所以必须要求测量结果可以溯源至所定义的标准,如SI单位、标准物质或(如果适用)所定义的方法或经验方法(参见5.
2节).
内部质量控制程序、水平测试和实验室认可可以作为辅助方法来证明与给定标准的溯源性.
上述要求的结果是:化学家们就其所从事的分析工作,正受到越来越大的压力要求其证明其结果的质量,特别是通过度量结果的可信度来证明结果的适宜性.
这一般包括期望某个结果与其他结果相吻合的程度,通常与所使用的分析方法无关.
度量该项内容的一个有用的方法就是测量不确定度.
虽然化学家们认识测量不确定度的概念已经有很多年了,但是直到1993年ISO才联合BIPM、IEC、IFCC、IUPAC、IUPAP和OIML出版了《测量不确定度表述指南》[H.
2],该指南正式确定了适用于广泛测量领域的评估和表达测量不确定度的通用原则.
本指南文件说明了ISO指南的概念如何运用到化学测量中.
它首先引入了不确定度的概念及不确定度和误差的区别,然后描述了评估不确定度的步骤,并在附录A中给出了评估过程的实际例子.
CNAS-GL06:2006第5页共148页2006年06月01日发布2006年07月01日实施评估不确定度时,要求分析人员密切注意产生不确定度的所有可能来源.
虽然对不确定度来源的详尽研究需要付出相当多的工作,但是,所付出的努力与所分析对象的复杂程度应相适宜.
实际上,初步的分析就可快速确定不确定度最重要的来源.
正如实例中所显示的那样,合成不确定度的数值几乎完全取决于那些重要的不确定度分量.
评估不确定度时,正确的做法应该是集中精力分析最大的不确定度分量.
此外,对于某特定实验室中指定方法(即:特定的测量程序)完成不确定度评估后,经过有关质量控制数据验证后,这一不确定度估计值能可靠地适用于以后该实验室使用该方法所得到的结果中.
只要测量过程本身或所使用的设备未变化,就不需要再进一步进行不确定度评估了.
在测量过程本身或所使用的设备发生变化时,需要重新审查不确定度评估结果,并将这项工作作为通常进行的方法再确认的一部分.
EURACHEM第一版"分析测量中的不确定度评估"指南[H.
3]是根据ISO指南于1995年出版的.
编制EURACHEM指南的第二版时,依据了化学实验室测量不确定度评估的实践经验,并更清楚地认识到实验室引入正式质量保证程序的必要性.
第二版强调的是实验室引入测量不确定度评估程序时,应该与其现有的质量保证措施结合起来,因为上述质量保证措施通常提供了评估测量不确定度所需要的很多信息.
所以本指南中明确给出了完全按ISO指南中的原则使用确认和有关的数据来进行不确定度评估.
本方法也与ISO17025:1999[H.
1]的要求相吻合.
注:附录A中给出了实例.
附录B中给出了定义列表.
按惯例将文中第一次出现的有定义的术语用黑体字打印,并在方括号中给出了其在附录B中的索引号.
定义主要摘自国际计量学基本和通用标准术语词汇表(VIM)[H.
4]、指南[H.
2]和ISO3534(统计学-词汇表和符号)[H.
5].
附录C中用通用术语列出了产生测量结果的化学分析的总体结构.
附录D描述了用于识别不确定度分量和筹划下一步所需试验的通用程序.
附录E中列出了分析化学中测量不确定度评估所使用的某些统计方法.
附录F中讨论了接近检出限时测量不确定度的处理方法.
附录G列出了很多常见的不确定度来源和评估不确定度数值的方法.
附录H是参考文献.
CNAS-GL06:2006第6页共148页2006年06月01日发布2006年07月01日实施化学分析中不确定度的评估指南1.
目的与范围1.
1本指南给出了定量化学分析中评估和表述不确定度的详细指导.
它是基于"ISO测量不确定度表述指南"[H.
2]中所采用的方法,适用于各种准确度和所有领域-从日常分析到基础研究、到经验方法和合理方法(参见第5.
3节).
需要化学测量并可以使用本指南原理的一些常见领域有:·制造业中的质量控制和质量保证;·判定是否符合法定要求的测试;·使用公认方法的测试;·标准和设备的校准;·与标准物质研制和认证有关的测量活动;·研究和开发活动.
1.
2请注意在某些情况下可能还需要额外的指南.
特别是,本指南中未包括如何使用公认方法(包括多重测量方法)给标准物质赋值,在符合性声明中如何使用不确定度估计值和在低浓度如何使用和表述不确定度,对此可能还需要额外的指南.
与取样操作有关的不确定度也未在本指南中明确涉及.
1.
3由于一些领域的实验室已采用了正式的质量保证措施,所以本指南第二版说明了应该如何使用从下列过程获得的数据进行测量不确定度评估:·某一实验室作为规定测量程序[B.
8]使用某种方法,对该方法所得分析结果的已识别来源的不确定度影响的评价;·一个实验室中规定的内部质量控制程序的结果;·为了确认分析方法而在一些有能力的实验室间进行的协同试验的结果;·用于评价实验室分析能力的水平测试项目的结果.
1.
4、无论是进行测试还是评估测量程序的操作性能,本指南始终认为有效的质量保证和控制措施,可以确保过程稳定并受控.
这些措施通常包括:诸如合格的工作人员、对设备和试剂的正确维护和校准、使用适当的参考标准、文件化的测量程序、使用适当的核查标准和控制图.
参考文献[H.
6]中有关于分析质量保证程序的更进一步的信息.
注:本段的意思是本指南认为所有分析方法的实施均是按已充分文件化的程序来进行.
因此对分CNAS-GL06:2006第7页共148页2006年06月01日发布2006年07月01日实施析方法的任何引用都意味着其有这类的程序.
严格意义上讲,测量不确定度仅适用于这类程序的结果,而不是通常的测量方法[B.
9].
2.
不确定度2.
1不确定度的定义2.
1.
1本文所使用的(测量)不确定度的术语定义取自现行版本的《国际计量学基本和通用术语词汇表》[H.
4].
定义如下:测量不确定度:表征合理地赋予被测量之值的分散性,与测量结果相联系的参数.
注1:这个参数可能是,如标准偏差【B.
23】(或其指定倍数)或置信区间宽度.
注2:测量不确定度一般包括很多分量.
其中一些分量是由测量序列结果的统计学分布得出的,可表示为标准偏差.
另一些分量是由根据经验和其他信息确定的概率分布得出的,也可以用标准偏差表示.
在ISO指南中将这些不同种类的分量分别划分为A类评定和B类评定.
2.
1.
2在化学分析的很多情况中,被测量【B.
6】是某被分析物的浓度P*P.
然而,化学分析也可用于测量其他量,例如颜色、pH值等,所以本文中使用了"被测量"这一通用术语.
*在本指南中,未加限定词的术语"浓度"适用于任何具体的量,如质量浓度、数量浓度、数字浓度或体积浓度,除非引用了单位(例如,用mglP-1P表示的浓度明显就是质量浓度).
也应注意,用来表示成份的其他量,如质量分数、物质含量和摩尔分数,能直接表示浓度.
2.
1.
3上述不确定度的定义主要考虑了分析人员确信被测量可以被合理地赋值的数值范围.
2.
1.
4通常意义上,不确定度这一词汇与怀疑一词的概念接近.
在本指南中,如未加限定词,不确定度一词可能指上述定义中的有关参数,或是指对于一个特定量的有限知识.
测量不确定度一词没有对测量有效性怀疑的意思,正相反,对不确定度的了解表明对测量结果有效性的信心增加了.
2.
2不确定度的来源2.
2.
1在实际工作中,结果的不确定度可能有很多来源,例如定义不完整、取样、基体效应和干扰、环境条件、质量和容量仪器的不确定度、参考值、测量方法和程序中的估计和假定、以及随机变化等(在第6.
7节中给出了不确定度来源更完整的说明).
2.
3不确定度的分量CNAS-GL06:2006第8页共148页2006年06月01日发布2006年07月01日实施2.
3.
1在评估总不确定度时,可能有必要分析不确定度的每一个来源并分别处理,以确定其对总不确定度的贡献.
每一个贡献量即为一个不确定度分量.
当用标准偏差表示时,测量不确定度分量称为标准不确定度【B.
13】.
如果各分量间存在相关性,在确定协方差时必须加以考虑.
但是,通常可以评价几个分量的综合效应,这可以减少评估不确定度的总工作量,并且如果综合考虑的几个不确定度分量是相关的,也无需再另外考虑其相关性了.
2.
3.
2对于测量结果y,其总不确定度称为合成标准不确定度【B.
14】,记做uBcB(y),是一个标准偏差估计值,它等于运用不确定度传播律将所有测量不确定度分量(无论是如何评价的)合成为总体方差的正平方根(请参考第8节).
2.
3.
3在分析化学中,很多情况下要用到扩展不确定度【B.
15】U.
扩展不确定度是指被测量的值以一个较高的置信水平存在的区间宽度.
U是由合成标准不确定度uBcB(y)乘以包含因子【B.
16】k.
选择包含因子k时应根据所需要的置信水平.
对于大约95%的置信水平,k值为2.
注:对于包含因子k应加以说明,因为只有如此才能复原被测量值的合成标准不确定度,以备在可能需要用该量进行其他测量结果的合成不确定度计算时使用.
2.
4误差和不确定度2.
4.
1区分误差和不确定度很重要.
误差【B.
19】定义为被测量的单个结果和真值【B.
3】之差.
所以,误差是一个单个数值.
原则上已知误差的数值可以用来修正结果.
注:误差是一个理想的概念,不可能被确切地知道.
2.
4.
2另一方面,不确定度是以一个区间的形式表示,如果是为一个分析过程和所规定样品类型做评估时,可适用于其所描述的所有测量值.
一般不能用不确定度数值来修正测量结果.
2.
4.
3此外,误差和不确定度的差别还表现在:修正后的分析结果可能非常接近于被测量的数值,因此误差可以忽略.
但是,不确定度可能还是很大,因为分析人员对于测量结果的接近程度没有把握.
2.
4.
4测量结果的不确定度并不可以解释为代表了误差本身或经修正后的残余误差.
2.
4.
5通常认为误差含有两个分量,分别称为随机分量和系统分量;2.
4.
6随机误差【B.
20】通常产生于影响量的不可预测的变化.
这些随机效应使得被测量的重复观察的结果产生变化.
分析结果的随机误差不可消除,但是通常可以通过增加观察CNAS-GL06:2006第9页共148页2006年06月01日发布2006年07月01日实施的次数加以减少.
注1:虽然在一些不确定度的出版物中是这样说的,但是,实际上算术平均值[B.
22]或一系列观察值的平均值的实验标准差不是平均值的随机误差.
它是由一些随机效应产生的平均值不确定度的度量.
由这些随机效应产生的平均值的随机误差的准确值是不可知的.
2.
4.
7系统误差【B.
21】定义为在对于同一被测量的大量分析过程中保持不变或以可以预测的方式变化的误差分量.
它是独立于测量次数的,因此不能在相同的测量条件下通过增加分析次数的办法使之减小.
2.
4.
8恒定的系统误差,例如定量分析中没有考虑到试剂空白,或多点设备校准中的不准确性,在给定的测量值水平上是恒定的,但是也可能随着不同测量值的水平而发生变化.
2.
4.
9在一系列分析中,影响因素在量上发生了系统的变化,例如由于试验条件控制得不充分所引起的,会产生不恒定的系统误差.
例子:1、在进行化学分析时,一组样品的温度在逐渐升高,可能会导致结果的渐变.
2、在整个试验的过程中,传感器和探针可能存在老化影响,也可能引入不恒定的系统误差.
2.
4.
10测量结果的所有已识别的显著的系统影响都应修正.
注:测量仪器和系统通常需要使用测量标准或标准物质来调节或校准,以修正系统影响.
与这些测量标准或标准物质有关的不确定度及修正过程中存在的不确定度必须加以考虑.
2.
4.
11误差的另一个形式是假误差或过错误差.
这种类型的误差使测量无效,它通常由人为失误或仪器失效产生.
记录数据时数字进位、光谱仪流通池中存在的气泡、或试样之间偶然的交叉污染等原因是这类误差的常见例子.
2.
4.
12有此类误差的测量是不可接受的,不可将此类误差合成进统计分析中.
然而,因数字进位产生的误差可进行修正(准确),特别是当这种误差发生在首位数字时.
2.
4.
13假误差并不总是很明显的.
当重复测量的次数足够多时,通常应采用异常值检验的方法检查这组数据中是否存在可疑的数据.
所有异常值检验中的阳性结果都应该小心对待,可能时,应向实验者核实.
通常情况下,不能仅根据统计结果就剔除某一数值.
2.
4.
14使用本指南获得的不确定度并没有考虑出现假误差或过错误差的可能性.
3.
分析测量和不确定度CNAS-GL06:2006第10页共148页2006年06月01日发布2006年07月01日实施3.
1方法确认3.
1.
1在实践中,日常检测使用的分析方法的适用性通常是通过方法确认研究【H.
7】加以评价的.
这种研究对于整体性能和个别影响因子分别产生数据,并可以在正常使用中利用这些数据来评估该方法产生的测量结果的测量不确定度.
3.
1.
2方法确认研究依赖于整体方法性能参数的确定.
这些参数是在方法开发和实验室间研究或按照实验室内确认方案获得的.
单个的误差或不确定度来源通常仅当其与所使用的总体精密度测量相比较时比较显著才加以研究.
重点主要放在识别和消除(而不是修正)显著影响.
这就形成了大多数潜在的显著影响因素已被识别,并已通过与整体精密度相比较来检查其显著性,并表明可以忽略的局面.
在这种情况下,分析人员可以得到的数据主要包括总体性能数据及很多影响是非显著的证据和残余的某些显著影响的测量值.
3.
1.
3定量分析方法的确认研究通常确定了下列部分或全部参数:U精密度U:主要精密度测量包括重复性标准偏差sBrB、复现性标准偏差sBRB,(ISO3534-1)和中间精密度,有时表示为sBZiB,其中i表示变化因素的数目(ISO5725-3:1994).
重复性sBrB表示在短时间内由同一操作人员操作同一设备等在同一间实验室内观察到的变异性.
sBrB可以在一个实验室内评估,也可以通过实验室间研究来评估.
特定方法的实验室间的复现性标准偏差sBRB可能只能用实验室间研究的方法直接评估;它表示不同实验室分析同一样品的差异性.
当实验室内的一个或几个因素(例如时间、设备和操作人员)发生变化时,中间精密度与所观察到的结果的变化有关;所得到的数值可能有所不同,这取决于那些因素是保持恒定的.
中间精密度评估通常是在实验室内获得的,但是也可以由实验室间研究取得.
无论是通过独立方差合成的方法得到的,还是通过研究完整的操作方法获得的,分析过程所观察到的精密度是总体不确定度的基本分量.
U偏差U:分析方法的偏差通常是通过研究相关标准物质或通过加料研究而确定的.
使用适当的标准数值来确定总体偏差,在建立对公认的标准的溯源性【B.
12】时很重要(见3.
2节).
偏差可以表示为分析回收率(观察值除以期望值).
偏差应小到可忽略或应加以修正,但不论何种情况,与偏差确定有关的不确定度始终是总不确定度的一个基本分量.
U线性U:线性是分析方法在测量一个区间内的浓度时的重要特性.
对于纯标准或实际样品响应能否成线性是可以确定的.
通常并不将线性量化,而是通过检验或使用非线性显著性检测来检查线性.
显著的非线性通常使用非线性校准函数加以修正,或通过选择更严格CNAS-GL06:2006第11页共148页2006年06月01日发布2006年07月01日实施的操作区间加以消除.
残余的线性偏差通常是在计算覆盖几个浓度的总体精密度估计值或与校准有关的不确定度中充分考虑(附录E.
3).
U检出限U:在方法确认中,通常确定检出限只是为了建立一个方法实际操作区间的下限.
虽然接近检出限的不确定度在评估时需要仔细考虑和特别处理(附录F),检出限(无论它是如何确定的)与测量不确定度的评估没有直接关系.
U稳健性U:很多方法开发和方法确认方案中需要直接研究特定参数的灵敏度.
通常通过初步的'稳健性测试'来得到,即观察一个和多个参数变化的影响.
如果影响显著(与稳健性测试的精密度相比较),就需要进一步详细研究来确定影响的大小,并因此选择一个所允许的操作区间.
因此稳健性测试的数据也可提供关于重要参数影响的信息.
U选择性/特定性U:虽然定义不是很严格,但是这两个术语均指一个分析方法对所要求的被分析物的专一响应程度.
典型的选择性研究中需要研究可能的干扰物的影响,通常采用在空白和被分析物加强型的样品中加入潜在的干扰物并观察其响应值.
结果通常用于证明实际影响并不显著.
然而,因为这种研究是直接测量响应值的变化,所以当干扰物的浓度范围已知时,有可能使用这些数据来评估与潜在的干扰物有关的不确定度.
3.
2方法性能的实验研究3.
2.
1方法确认和方法性能研究的详细设计和实施在别处有详细的讨论【H.
7】,在此不再重复.
然而,研究中影响不确定度评估的主要原则是很关键的,因此在下面加以讨论.
3.
2.
2代表性是最重要的.
也就是说,研究应尽可能反映方法正常使用中影响因素数量和范围的实际情况,并涵盖方法范畴内的浓度范围和样品类型.
例如,在精密度试验中如果一个因子已代表性地变化,那么这个因子的影响就可以直接出现在所观察到的变化中,而不需要做额外研究,除非方法需要进一步的优化.
3.
2.
3本文中,代表性变化是指影响参数必须具有与该参数的不确定度相适应的数值分布.
对于连续参数,这可以是允许的范围或给出的不确定度;对于样品基体等非连续因子,这个范围与方法正常使用中所允许或者所遇到的不同类型相一致.
注意代表性不只扩展到了数值的范围,而且包括其分布.
3.
2.
4在选择变化因子时,必须保证只要可能就改变那些较大的影响量.
例如,日与日之间的变化(可能产生于重新校准的影响)比重复性更大时,5天中的每一天进行2次测量就比2天中的每一天进行5次测量要给出更好的中间精密度估计值.
在充分的控制下,在不CNAS-GL06:2006第12页共148页2006年06月01日发布2006年07月01日实施同的天里进行10次单独的测量可能会更好,虽然它对于日内重复性没有提供额外的信息.
3.
2.
5通常处理随机选取的数据比处理从系统变化获取的数据更简单.
例如:在足够长的时间内进行随机次数的试验通常包括代表性的环境温度影响,而按照24小时间隔系统完成的试验可能由于工作日内常规的环境温度变化而产生偏差.
前者只需要评估总体标准偏差,后者还需要环境温度的系统变化,并随后按照温度的实际分布进行调整.
然而随机变化有效性降低了.
小量的系统研究就可以快速确定一个影响量的大小,但是通常需要测量30多次才能给出高于约20%相对准确度的不确定度分量.
只要可能,系统地研究少量的主要影响因素是更好地选择.
3.
2.
6当因素已知或可能产生交互作用时,必须保证交互作用的影响加以考虑了.
这可以通过保证随机选取不同水平的交互作用参数或通过以获得方差和协方差信息为目的缜密的系统设计来达到.
3.
2.
7在对总体偏差研究时,标准物质和数值应与日常检测的物质相关.
3.
2.
8为调查和确定某个影响量的显著性而进行的研究,必须有足够的力量在这些影响量变得实际显著之前确定这些影响量.
3.
3溯源性3.
3.
1能够可信地比较来自不同实验室的结果或同一实验室不同时期的结果是重要的.
通过保证所有的实验室均使用同样的测量尺度或同样的'参考点',就可达到这一点.
许多情况下是通过建立能够到达国家或国际基准,理想的情况下是测量国际单位制SI(为了长期的一致)的校准链.
熟悉的例子是分析天平.
每个天平用标准砝码来校准,而后者(最终)跟国家基准核对,如此直至千克基准.
这种可到达已知参考值的不间断链的比较提供了对共同'参考点'的溯源性,确保不同的操作者使用同一测量单位.
在日常测量中,对用来获得或控制某个测量结果的所有的中间测量,均建立溯源性,可极大地帮助达到一个实验室(一个时期)和另一个实验室(另一个时期)的测量结果的一致性.
因此在所有测量领域中溯源性是一个重要的概念.
3.
3.
2溯源性的正式定义[H.
4]是:"通过一条具有规定不确定度的不间断的比较链,使测量结果或测量标准的值能够与规定的参考标准,通常是与国家测量标准或国际测量标准联系起来的特性".
需要提及不确定度是由于实验室间的一致性在一定程度上受到每个实验室的溯源性CNAS-GL06:2006第13页共148页2006年06月01日发布2006年07月01日实施链所带来的不确定度的限制.
溯源性因此与不确定度紧密联系.
溯源性提供了一种将所有有关的测量放在同一测量尺度上的方法,而不确定度则表征了校准链链环的'强度'以及从事同类测量的实验室间所期望的一致性.
3.
3.
3通常,某个可溯源至特定参考标准的结果的不确定度,将由该标准的不确定度与对照该标准所进行的测量的不确定度组成.
3.
3.
4完整的分析过程的结果的溯源性应通过下列步骤的综合使用来建立:1.
使用可溯源标准来校准测量仪器;2.
通过使用基准方法或与基准方法的结果比较;3.
使用纯物质的标准物质RM;4.
使用含有合适基体的有证标准物质CRM;5.
使用公认的、规定严谨的程序.
下面依次讨论每个步骤.
3.
3.
5测量仪器的校准在任何情况下,所使用的测量仪器的校准必须可溯源到适当的标准.
分析过程的定量阶段通常使用其值可溯源至SI的纯物质的标准物质来进行校准.
这种做法为这部分过程结果提供了至SI的溯源性.
然而,有必要使用额外的程序,为诸如萃取和样品净化等属于定量阶段之前的操作结果建立溯源性.
3.
3.
6使用基准方法来进行测量下面是目前对基准方法的描述:"测量的基准方法是一种具有最高计量学特性,其操作可用SI单位进行完整地描述并被理解,其结果无需参考相同量的标准即可接受的方法".
基准方法的结果通常直接可溯源至SI单位,并且相对于该参考标准具有所能获得的最小不确定度.
基准方法通常只由国家测量机构来实施,很少用于日常测试或校准.
如有可能,通过直接比较基准方法和测试或校准方法的测量结果来达到对基准方法结果的溯源性.
3.
3.
7使用纯物质标准物质(RM)通过测量含有或由已知量纯物质组成的样品可证明溯源性.
例如,可通过加料或标准加样来达到.
然而,评估测量系统对所使用的标准和所测试的样品的不同响应总是必要的.
可惜的是,对于许多化学分析,在加料或标准加样的具体例子中,对不同响应的修正和其CNAS-GL06:2006第14页共148页2006年06月01日发布2006年07月01日实施不确定度可能较大.
因此,虽然原则上该结果对SI单位的溯源性得以建立,但实际上,除最简单例子外,所有的例子,其结果的不确定度可能大得不可接受或甚至无法量化.
假如其不确定度不能量化,则其溯源性并未建立起来.
3.
3.
8使用有证标准物质(CRM)进行测量对有证基体的CRM进行测量,并将测量结果与其有证数值比较可证明溯源性.
当有合适基体的CRM时,该程序与使用纯物质RM相比能够降低不确定度.
假如CRM的值可溯源至SI,则这些测量也可溯源至SI单位,使用标准物质的不确定度评估在7.
5节讨论.
然而,即使在这种情况下,假如样品和标准物质的成分之间没有很好匹配,其结果的不确定度可能大得无法接受或甚至无法量化.
3.
3.
9使用公认程序通过使用规定严谨并且普遍接受的程序可达到适当的可比性.
该程序通常用输入参数加以规定,例如一组规定了的萃取时间,颗粒大小等等.
当这些输入参数的值按正常方式可溯源至所规定的参考标准时,使用这类程序产生的结果被认为是可溯源的.
其结果的不确定度来自所规定的输入参数的不确定度和规范不完整的影响以及执行过程中的变化(见7.
8.
1节).
当估计另一种方法或程序的结果可与这类公认程序的结果相比较时,则可通过比较两者的结果来建立对该公认值的溯源性.
4.
测量不确定度的评估过程4.
1不确定度的评估在原理上是很简单.
下述段落概述了为获取测量结果的不确定度估计值所要进行的工作.
随后的章节提供了用于不同情况下的附加指南,特别是关于使用方法确认研究的数据和使用正式的不确定度传播律.
这些步骤包括:第一步、规定被测量清楚地写明需要测量什么,包括被测量和被测量所依赖的输入量(例如被测数量、常数、校准标准值等)的关系.
只要可能,还应该包括对已知系统影响量的修正.
该技术规定资料应在有关的标准操作程序(SOP)或其他方法描述中给出.
第二步、识别不确定度的来源列出不确定度的可能来源.
包括步骤一所规定的关系式中所含参数的不确定度来源,但是也可以有其他的来源.
必须包括那些由化学假设所产生的不确定度来源.
附录D给出CNAS-GL06:2006第15页共148页2006年06月01日发布2006年07月01日实施了用结构图表示的一般步骤.
第三步、不确定度分量的量化测量或估计与所识别的每一个潜在的不确定度来源相关的不确定度分量的大小.
通常可能评估或确定与大量独立来源有关的不确定度的单个分量.
还有一点很重要的是要考虑数据是否足以反映所有的不确定度来源,计划其它的实验和研究来保证所有的不确定度来源都得到了充分的考虑.
第四步、计算合成不确定度在步骤三中得到的信息,是总不确定度的一些量化分量,它们可能与单个来源有关,也可能与几个不确定度来源的合成影响有关.
这些分量必须以标准偏差的形式表示,并根据有关规则进行合成,以得到合成标准不确定度.
应使用适当的包含因子来给出扩展不确定度.
图1用图示方法表示了该过程.
4.
2下面的章节对执行上述步骤提供了指南,并展示了如何根据所获得的有关多个不确定度来源的合成影响信息来简化这些步骤.
5.
第一步、被测量的技术规定5.
1在本不确定度评估的指南中,"被测量的技术规定"要求清楚明确地说明正在测量什么,并定量表述被测量的值与其所依赖的参数之间的关系.
这些参数可能是其他被测量、不能直接测量的量或者常数,并应明确过程中是否包括取样步骤.
如果有,与取样过程有关的不确定度评估必须考虑.
所有这类信息应在标准操作程序(SOP)中写明.
5.
2在分析测量中,特别重要地是要区别其测量结果独立于所使用的方法的测量和其测量结果依赖于所使用的方法的测量.
后者通常称作经验方法.
下面的例子将进一步澄清这一点.
例子:1.
测定合金中镍含量的方法通常会产生相同的结果,该结果以相同单位表示,一般表示为质量或摩尔分数.
原则上,需要对方法偏差或基体产生的任何系统影响进行修正,虽然更为常用的做法是确保任何此类影响是较小的.
除了作为参考信息外,结果通常无需引述所使用的特定方法.
该方法不是经验方法.
CNAS-GL06:2006第16页共148页2006年06月01日发布2006年07月01日实施图1不确定度评估过程第一步第二步第三步第四步开始规定被测量识别不确定度来源将现有数据的不确定度来源分组以简化评估量化分组分量量化其他分量将分量转换为标准偏差计算合成标准不确定度审定,如必要,重新评估较大的分量计算扩展不确定度结束CNAS-GL06:2006第17页共148页2006年06月01日发布2006年07月01日实施2.
"可萃取脂肪"的测定可能相差很大,它取决于所规定的萃取条件.
由于"可萃取脂肪"完全取决于条件的选择,所以所使用的方法就是经验方法.
考虑对该方法的内在偏差进行修正是无意义的,因为被测量是由所使用的方法所定义的.
结果的报告通常提到所使用的方法,而不对该方法的内在偏差进行修正.
该方法被认为是经验方法.
3.
当基质或基体的变化具有很大的和不可预测的影响时,通常开发程序的唯一目的是达到测量同一材料的实验室之间具有可比性.
然后可将该程序采用为地方,国家或国际标准方法,凭此进行贸易或做出决定,而无需绝对度量被分析物和实际含量.
按常规可不对方法偏差或基体影响进行修正(而不管在方法开发过程中它们是否已被最小化).
通常报告结果时没有对基体或方法偏差进行修正.
该方法被认为是经验方法.
5.
3经验方法和非经验方法(有时称为合理方法)的区别很重要,因为它影响了不确定度的评估.
在上述例子2和例子3中,因为采用了习惯做法,与一些较大的影响量相关联的不确定度在日常使用中并不相关.
所以,应适当考虑结果是独立于还是依赖于所使用的方法,并且只有那些与所报告的结果有关的影响量才应包括在不确定度评估过程中.
6.
第二步识别不确定度来源6.
1应列出不确定度有关来源的完整清单.
在本步骤,无需考虑单个分量的量化问题.
本步骤的目的是明确应考虑什么.
在第三步中,将考虑处理每一个来源的最佳方法.
6.
2在列出不确定度来源的清单时,通常方便的办法是从那些根据中间数值计算被测量的基本表达式开始.
这个表达式中的所有参数可能都有一个与其数值相关的不确定度,因此都是不确定度的潜在来源.
此外,也可能有其他参数并没有明显地出现在用于计算被测量数值的表达式中,但却影响该测量结果,例如:萃取时间或温度.
它们也是不确定度的潜在来源.
所有这些不同的来源都应包括在内.
在附录C(分析过程的不确定度)中给出了其他信息.
6.
3附录D中的因果图是列出不确定度来源的一种非常方便的方法,它表示了他们之间的相互关系,以及他们对结果的不确定度的影响.
它也有助于避免重复计算不确定度来源.
虽然还有其他方法可以列出不确定度的来源,本书的后面章节和附录A的例子中均采用因果图的方法.
其他信息参见附录D(分析不确定度来源).
6.
4列出不确定度的来源后,他们对于测量结果的影响原则上可以用一个正式的测量模型CNAS-GL06:2006第18页共148页2006年06月01日发布2006年07月01日实施来表述,其中每一个影响量都与公式中的一个参数有关或在公式中是变量.
该公式然后形成了用影响该测量结果的所有独立因素表示的测量过程的完整模型.
该函数可能非常复杂,不能明确地写出,但是只要可能,就应该这样做.
因为表达式的方式通常决定了合成各个不确定度分量的方法.
6.
5还有一个很有用地技巧是将测量过程考虑成一系列独立的操作(有时称为单元操作),每一个操作可以单独评价以得到与之相关地不确定度估计值.
当类似的测量过程共享相同地单元操作时,这是一个很有用地方法.
每个操作各自的不确定度构成了总不确定度的分量.
6.
6实际上,在分析测量中,更普遍的做法是考虑与整体方法性能的要素有关的不确定度,例如可观察的精密度和相对于合适的标准物质所测得的偏差.
这些构成了不确定度评估中的主要分量,最好在模型中表示为影响结果的独立因素.
然后,有必要对其他可能的分量进行评估,只需要检查他们是否显著,并只量化那些显著的量.
尤其适用于利用方法确认数据的本方法的进一步指南见第7.
2.
1节.
6.
7典型的不确定度来源包括:·U取样当内部或外部取样是规定程序的组成部分时,例如不同样品间的随机变化以及取样程序存在的潜在偏差等影响因素构成了影响最终结果的不确定度分量.
·U存储条件当测试样品在分析前要储存一段时间,则存储条件可能影响结果.
存储时间以及存储条件因此也被认为是不确定度来源.
·U仪器的影响仪器影响可包括,如对分析天平校准的准确度限制;保持平均温度的控温器偏离(在规范范围内)其设定的指示点,受进位影响的自动分析仪.
·U试剂纯度即使母材料已经化验过,因为化验过程中存在着某些不确定度,其滴定溶液浓度将不能准确知道.
例如许多有机染料,不是100%的纯度,可能含有异构体和无机盐.
对于这类物质的纯度,制造商通常只标明不低于规定值.
关于纯度水平的假设将会引进一个不确定度分量.
·U假设的化学反应定量关系CNAS-GL06:2006第19页共148页2006年06月01日发布2006年07月01日实施当假定分析过程按照特定的化学反应定量关系进行的,可能有必要考虑偏离所预期的化学反应定量关系,或反应的不完全或副反应.
·U测量条件例如,容量玻璃仪器可能在与校准温度不同的环境温度下使用.
总的温度影响应加以修正,但是液体和玻璃温度的不确定度应加以考虑.
同样,当材料对湿度的可能变化敏感时,湿度也是重要的.
·U样品的影响复杂基体的被分析物的回收率或仪器的响应可能受基体成份的影响.
被分析物的物种会使这一影响变得更复杂.
由于改变的热力情况或光分解影响,样品/被分析物的稳定性在分析过程中可能会发生变化.
当用'加料样品'用来估计回收率时,样品中的被分析物的回收率可能与加料样品的回收率不同,因而引进了需要加以考虑的不确定度.
·U计算影响选择校准模型,例如对曲线的响应用直线校准,会导致较差的拟合,因此引入较大的不确定度.
修约能导致最终结果的不准确.
因为这些是很少能预知的,有必要考虑不确定度.
·U空白修正空白修正的值和适宜性都会有不确定度.
在痕量分析中尤为重要.
·U操作人员的影响可能总是将仪表或刻度的读数读高或低.
可能对方法作出稍微不同的解释.
·U随机影响在所有测量中都有随机影响产生的不确定度.
该项应做为一个不确定度来源包括在列表中.
注:这些来源不一定是独立的.
7.
第三步量化不确定度CNAS-GL06:2006第20页共148页2006年06月01日发布2006年07月01日实施7.
1引言7.
1.
1已按第二步(第六章)的说明识别不确定度来源后,下一个步骤就是要量化这些不确定度来源所产生的不确定度.
可以通过以下方法:·评价每个不确定度来源的不确定度,然后将其按照第八章所述的方法合成.
例子A1至A3说明了这种方法的使用;或者·使用方法性能数据,直接确定来自一些或所有不确定度来源的结果的不确定度合成分量.
例子A4至A6给出了该过程的使用.
实践中,上述方法的组合使用通常是必要和便利的.
7.
1.
2不管使用哪种方法,评价不确定度最需要的大部分信息可从确认研究的结果、质量保证/质量控制(QA/QC)的数据和其他为核查方法性能进行的实验工作中得到.
然而,不是所有来源的不确定度都有评估其不确定度所需的数据,可能有必要进行7.
10至7.
14节所描述的其他工作.
7.
2不确定度的评估程序7.
2.
1用来评价总不确定度的程序取决于可提供的方法性能数据.
制定这一程序的步骤包括:·解决现有数据与信息需求之间的矛盾.
首先,检查不确定度来源表,看哪些不确定度来源可用现有的数据计算,而不管现有的数据是来自对具体分量的明确研究,还是来自整个方法实验过程中的隐含变化.
应对照步骤二所列的表来核查这些来源,并列出任何余下的来源,以提供哪些不确定度分量已包括在内的可审查记录.
·策划获取所需的其它数据对于没有被现有数据适当地包括的不确定度来源,可以从文献或现有资料(证书、仪器规格等)中获取其它信息,或策划实验以获取所需其它数据.
附加的实验可采取具体研究单个不确定度分量的方式,或采用常用的方法性能研究以保证重要因素有代表性的变化.
7.
2.
2很重要的是认识到不是所有的分量都会对合成不确定度构成显著的贡献.
实际上,只有很少的分量才会有显著影响.
除非数量很多,比最大的分量小三分之一的那些分量无需深入评估.
对于每一个分量或合成分量的贡献进行初步评估,去掉那些不重要的分量.
CNAS-GL06:2006第21页共148页2006年06月01日发布2006年07月01日实施7.
2.
3下面的章节对将要采取的程序提供了指南,这取决于现有的数据和所需的其它信息.
第7.
3节给出了使用以前的实验研究数据(包括确认数据)的要求.
第7.
4节简述了单纯从单个不确定度来源评估不确定度的方法.
对于所识别的所有或很少不确定度来源来说这可能是必要的,这取决于现有的数据,因此也在稍后的章节中加以考虑.
第7.
5直至7.
9节描述了在不同情况下评估不确定度的方法.
7.
5节适用于使用极为匹配的标准物质的情况.
7.
6节适用于使用协同研究数据,7.
7节适用于使用实验室内部确认数据.
7.
8节描述了经验方法的特殊处理.
7.
9节讨论了特别方法(ad-hocmethod).
量化单个不确定度分量的方法,包括实验研究、文献和其他数据、建立模型和专业判断,详见第7.
10到7.
14节.
7.
15节讨论了在不确定度评估中对于已知偏差的处理.
7.
3以前研究的相关性7.
3.
1当不确定度评估至少部分是基于以前的方法性能研究时,有必要证明使用以前研究结果的有效性.
典型地包括:·证明以前所得到的精密度具有相当水平.
·特别是通过用相关标准物质偏差的确定(例,见ISOGuide33[H.
8]),通过适当的加料研究或通过有关水平测试项目或其他实验室比对的满意表现来证明使用以前所得到的偏差数据的合理性.
·通过定期质量控制样品结果表明持续运行在统计控制之内以及实施有效的分析质量保证程序.
7.
3.
2满足上述条件,并且方法在其使用范围和领域内应用时,通常可以使用该实验室以前的研究(包括确认研究)数据来直接评估该实验室的不确定度.
7.
4量化单个分量来评估不确定度7.
4.
1有些情况下,特别是没有或很少有方法性能数据时,最适合的程序可能是分别评估每个不确定度分量.
7.
4.
2合成单个分量的一般程序是准备一个详细的试验过程的定量模型(参考第5节和第6节,尤其是第6.
4节),评估与单个输入参数相关的标准不确定度,并使用不确定度传播律对其进行合成,参见第8节.
7.
4.
3为了更清楚的说明问题,通过试验方法和其他方法评估单个分量的详细指南推迟到第7.
10到7.
14节中给出.
附录A中的例子A1和A3给出了该程序的详细描述.
使用该程序的CNAS-GL06:2006第22页共148页2006年06月01日发布2006年07月01日实施详细指南也参见ISO指南【H2】.
7.
5极匹配的有证标准物质7.
5.
1对有证标准物质的测量通常作为方法确认或重新确认的组成部分,有效地达到了对整个测量程序用可追溯的标准物质所进行的校准.
因为这一程序提供了许多不确定度潜在的来源的综合影响的信息,所以它为不确定度评估提供了很好的数据.
详见7.
7.
4节.
注:ISO指南33[H.
8]对于使用标准物质检查方法性能方面给出了有用的描述.
7.
6使用以前的协同方法开发和确认研究数据来评估不确定度7.
6.
1为确认公开发表的方法所做的协同研究,例如根据AOAC/IUPAC协议[H.
9]或者ISO5725标准[H.
10],是支持不确定度评估的有价值的数据来源.
这些数据通常包括对于不同水平的响应时复现性标准偏差的估计值sBRB,sBRB对响应水平相关性的线性估计值、以及可能还包括基于有证标准物质CRM研究的偏差估计值.
如何使用这个数据,取决于进行该项研究时考虑了哪些因素.
在上述"解决矛盾"阶段(7.
2节),有必要识别那些没有包括在协同研究数据内的不确定度来源.
需加以特别关注的来源包括:·U取样U:协同研究很少涉及取样步骤.
如果实验室内所使用的方法涉及到了二级取样,或被测量(见技术规定)是从一个小样品来估计整批产品的特性,必须研究取样的影响,并将这些因素包括在内.
·U预处理U:很多研究中,样品是均质的,分发前还可能需要进一步稳定.
可能有必要调查并增加实验室内使用该特定的预处理程序的影响.
·U方法偏差U:方法偏差的检查通常在实验室间研究之前或之中进行,只要可能就通过比较参考方法或标准物质进行.
当偏差自身、所使用的参考数值的不确定度和与偏差检查有关的精密度,比起sBRB来均小时,不必额外考虑偏差不确定度的影响.
否则,就必须考虑.
·U条件的变化U:参加研究的实验室可能倾向于采取所允许的实验条件范围的中间值,这导致低估了方法定义中可能的结果范围.
这些影响经研究表明对于其全部允许的范围内不显著,可以不考虑.
·U样品基体的变化U:基体成份或干扰物水平超出了研究范围时,由此引入的不确定度需要加以考虑.
7.
6.
2每一个没有被共同研究数据所包括的重要的不确定度来源都应以标准不确定度的形CNAS-GL06:2006第23页共148页2006年06月01日发布2006年07月01日实施式加以评估,并按通常方式与重现性标准偏差sBRB合成(第8节).
7.
6.
3对于在定义范围内操作的方法,当"解决矛盾"阶段表明所有已识别的不确定度来源已包括在确认研究中,或来自诸如7.
6.
1节讨论的任何残余来源的贡献经证明可以忽略不计时,则重现性标准偏差sBR,,B必要时对浓度进行调整,可以作为合成标准不确定度使用.
7.
6.
4本程序使用的例子见A6(附录A).
7.
7使用实验室内开发和确认研究进行不确定度评估7.
7.
1实验室内开发和确认研究主要包括3.
1.
3节所指的方法性能参数的确定.
从这些参数进行不确定度评估使用下面数据:·现有的最佳总精密度估计值.
·现有的最佳总体偏差及其不确定度估计值.
·对与在上述整体性能研究中未能完整得到考虑的影响量有关的不确定度的量化.
精密度研究7.
7.
2精密度的评估应尽可能在一段较长的时间内,并特意使影响结果的所有因素自然变化.
可以通过下述得到:·一段时间内对典型样品的几次分析的结果的标准偏差,应尽可能由不同的分析人员操作和使用不同的设备(质量控制核查样品的测量结果能提供这方面的信息).
·对于多个样品中的每一个均进行重复分析所得的标准偏差.
注:重复分析应在有实质不同的时间内进行以获得中间精密度.
同一批内进行的重复分析只能提供重复性的估计值.
·从正式的多因素实验设计,用ANOVA加以分析,为每一个因素提供单独的方差估计值.
7.
7.
3注意精密度经常随响应水平的不同而产生大的变化.
例如,所观察到的标准偏差经常随着被分析物的浓度显著和系统地增加.
在这些情况下,不确定度评估值应进行调整以考虑适用于特定结果的精密度.
附录E4中给出了处理与响应水平有关的不确定度分量的指南.
偏差研究7.
7.
4总体偏差最好通过使用完整的测量程序对有关有证标准物质进行重复分析来估计.
经分析,所得到的偏差如果不显著,与此偏差相关的不确定度就是有证标准物质数值的标CNAS-GL06:2006第24页共148页2006年06月01日发布2006年07月01日实施准不确定度和与偏差有关的标准偏差的简单合成.
注:用这做方式估计的偏差就是实验室操作偏差与所使用方法的内在偏差的合成.
当所使用的方法是经验方法时,要给予特别考虑,见7.
8.
1节.
·当标准物质只大致代表测试样品时,应考虑其他因素,包括(适用时):成份和均质性的差别;标准物质通常比测试样品更均质.
必要时,由专业判断给出的估计值应被用来给这些不确定度赋值(见7.
14节).
·被分析物的不同浓度产生的影响;例如,通常发现萃取的损失在被分析物的高浓度和低浓度是不同的.
7.
7.
5所研究方法的偏差也可以将其结果与参考方法的结果进行比较而得到.
如果结果表明偏差在统计学上是不显著的,标准不确定度就是参考方法的不确定度(如适用,见7.
8.
1节)与所测的两个方法之差的标准不确定度的合成.
后者的不确定度分量由用于决定该差别统计上是否显著的显著性试验中所用的标准偏差术语表示,见下面例子的说明.
例:决定硒浓度的某个方法(方法1)与参考方法(方法2)比较.
每一个方法的结果(用1mgkg表示)如下:xsn方法15.
401.
475方法24.
762.
755标准偏差合并后得合并标准偏差sBcB205.
2255)15(75.
2)15(47.
122=+*+*=cs以及相应的t值:46.
04.
164.
05151205.
2)76.
440.
5(==+=t对于自由度为8,tBcritB是2.
3,因此两个方法所给的结果的平均值无显著差别.
然而,该结果差(0.
64)是与上述1.
4标准偏差术语相比较的.
数值1.
4是与结果差有关的标准偏差,因此代表了与所测得的偏差相关的不确定度分量.
CNAS-GL06:2006第25页共148页2006年06月01日发布2006年07月01日实施7.
7.
6总体偏差也可以通过加被分析物至一种以前已研究过的物质中的方法来进行评估.
研究标准物质(上述)的考虑也同样适用.
此外,所加入物质和样品本身的物质的不同表现应加以考虑,并给出相应的允差.
此类允差可基于下述给出:·对于一定范围的基体和所加的被分析物水平,研究所观察到的偏差的分布.
·将对标准物质所观察到的结果与在同一标准物质中所加被分析物的回收率比较.
·基于具有已知极端表现的具体材料而所做的判断.
例如,在牡蛎组织,一个普通的海组织参考物,众所周知在消化时易与钙盐一起共同沉淀某些元素,可提供作为不确定度评估基础的'最不利情况'回收率的估计值(例如,将最不利情况作为矩形或三角形分布的端值).
·基于以前经验所做的判断.
7.
7.
7也可以通过比较特定方法和通过标准加入法测定的数值来评估偏差.
在标准加入法中,将已知量的被分析物加到测试样中,正确的被分析物浓度通过外推法得到.
与偏差有关的不确定度通常主要是外推法的不确定度,以及与(适合时)来自制备和添加贮存溶液的重要的不确定度分量一起合成.
注:为了直接相关,该加入应直接加到最初样品,而不是已制备的萃取物中.
7.
7.
8对于所有已知的和显著的系统影响必须进行校正,这是ISO指南中的通用要求.
当对一个显著的总体偏差进行修正时,与偏差有关的不确定度的评估按照7.
7.
5节所描述的有关处理不显著的偏差的情况进行.
7.
7.
9偏差显著时,但是又因为某个实际的原因忽略不计了,此时必须采取其它的措施(见7.
15节).
其他因素7.
7.
10剩下的其他因素的影响应分别估计.
可以通过实验变化的方法,也可通过已建理论预测的方法进行评估.
与这些因素相关的不确定度也应该加以评估、记录、并和其他分量按照常规方法合成.
7.
7.
11与该研究的精密度相比,这些剩余因素的影响被证明可以忽略不计时(即统计上非显著时),建议其不确定度分量等于与该因素有关的显著性检验的标准偏差.
例:对所允许的一个小时萃取时间变化的影响通过t-检验进行了研究,对于正常萃取时间和减少一个小时的萃取时间分别对同一样品测试了五次.
平均值和标准偏差(用mglP-1P表示)是:标准时间:平均值为1.
8,标准偏差为0.
21;而另一个时间:平均值为1.
7,标准偏差为0.
17.
t-检验使用合并方CNAS-GL06:2006第26页共148页2006年06月01日发布2006年07月01日实施差以获得t值,其中合并方差等于82.
05151037.
0)7.
18.
1(037.
0)15()15(17.
0)15(21.
0)15(22=+*==+*+*t与3.
2=critt相比,这是不显著的.
但注意该差值(0.
1)是与所计算的标准差术语来比较的,其值=12.
0)5/15/1(037.
0=+*.
该值就是与萃取时间允许变化的影响有关的不确定度分量.
7.
7.
12当测到某一个影响在统计学上是显著时,但是在实践中又足够小可以忽略不计时,参考7.
15节的方法处理.
7.
8经验方法的不确定度评估7.
8.
1'经验方法'是指在特定应用领域内,为测量比较的目的而一致同意使用的方法,其特征是被测量依赖于所使用的方法.
该方法因此定义了被测量.
例子包括从陶瓷中溶出金属的测定方法和食物中的食用纤维的测定方法(也见5.
2节和例A5).
7.
8.
2当此类方法用于其指定领域时,其方法偏差规定为零.
在这种情况下,偏差估计中应只和实验室的操作相关,不应额外考虑方法内在的偏差.
含义如下所述.
7.
8.
3无论是为了证明偏差可忽略还是为了测量偏差,进行标准物质研究时应使用经特定方法验证的标准物质或使用其值已该通过特定方法获得以供比较的标准物质.
7.
8.
4当具有此类特征的标准物质不能获得时,对偏差的整体控制就与对影响结果的方法参数控制相关,特别是时间、温度、质量、体积等因素.
与这些输入因素有关的不确定度因此必须进行评估,或证明其可以忽略不计,或对其定量化(见例子A6).
7.
8.
5经验方法通常作为协同研究对象,所以应按照7.
6节中方法评估其不确定度.
7.
9特别方法(ad-hocmethods)的不确定度评估7.
9.
1特别方法是在短期内或为一小批测试样而进行的探测性研究所建立的方法.
这种方法通常基于标准方法或实验室内制定的良好方法,但做了大的修改(例如研究不同的被分析物)并且通常没有合理的理由为所讨论的特定材料进行正式的确认研究.
7.
9.
2由于为建立有关不确定度分量所付出的努力有限,有必要大部分依赖于有关系统已知性能或这些系统内的有关部分的已知性能.
不确定度评估因此就应该基于一个有关系统CNAS-GL06:2006第27页共148页2006年06月01日发布2006年07月01日实施或多个系统的已知性能.
该性能信息必须被必要的研究所支持,以确认信息的相关性.
下面建议假定存在这样一个相关的系统,并且为获得可信的不确定度估计值已进行了充分的研究,或该方法包括了其他方法的部分,并且这些部分的不确定度以前已经确定.
7.
9.
3至少,有必要得到所讨论方法的总偏差的估计值和精密度的表示值.
测量偏差理想的做法是用标准物质,但实际上更常用的做法是通过加料回收率加以评估.
除了加料回收率要与相关系统内所观察的值相比较,以建立以前研究与所讨论的特别方法之间联系外,7.
7.
4节的讨论适用.
对所测试材料,在本研究要求内,特别方法所观察的总偏差,应与有关系统的观察值相当.
7.
9.
4最少的精密度试验由一次重复分析组成.
然而,我们推荐进行尽可能多的重复分析.
该精密度应与相关系统的精密度比较.
特别方法的标准偏差应相当的.
注:建议比较应基于检查.
统计上的显著性检验(如F-检验)对于重复次数小的重复分析通常不可靠,并且仅仅因为该检验的不充分将易于导致'不存在显著差异'的结论.
7.
9.
5当上述条件明确得到满足时,相关系统的不确定度评估值可以直接应用到特别方法所得的结果中,并根据浓度和其他已知因素进行适当调整.
7.
10单个分量的量化7.
10.
1几乎总是很有必要单独考虑一些不确定度的来源.
在有些情况下,只需考虑很少量的来源,在另一些情况下,特别是方法性能数据很少或没有时,应分别研究每个来源(见附录A的例1、2和3的说明).
有几个通用的方法来建立单个的不确定度分量:·输入变量的实验变化·根据现有数据,例如测量和校准证书·通过理论原则建立的模型·根据经验和假设模型的信息作出的判断这些不同的方法将分别简述如下.
7.
11单个不确定度分量的试验估计7.
11.
1通过针对特定参数的试验研究来进行不确定度分量的估计,通常是可能和可行的.
7.
11.
2随机影响的标准不确定度通常通过重复性试验测量,并以被测量值的标准偏差的形式定量表示.
实践中,通常重复试验不超过十五次,除非需要更高的精密度.
7.
11.
3其他典型的试验包括:CNAS-GL06:2006第28页共148页2006年06月01日发布2006年07月01日实施·单个参数变化对结果影响的研究.
尤其适合于独立于其他影响的连续、可控制参数的情况,如时间或温度.
结果随参数变化的变化率可通过实验数据获得.
然后将其直接与该参数的不确定度合成就可得到相关的不确定度分量.
注:与该研究现有精密度相比,参数的变化应该足够使结果产生大的变化.
(例如,重复测量的标准偏差的五倍).
·稳健性研究,系统地研究参数适当变化的显著性.
尤其适合于快速识别显著影响;并且通常用于方法优化.
该方法可用于离散影响的情况,如基体的变化,或小型仪器结构的变化,它们可能对结果产生不可预测的影响.
当发现某个因素是显著的,通常要进一步研究.
当不显著时,与之有关的不确定度是稳健性研究所得不确定度(至少对初步评估是这样).
·系统地多参数实验设计方案旨在评估因素影响和相互作用.
这类研究在研究绝对变量时尤其有用.
绝对变量是其值与影响量的大小无关的参数.
在一个研究中的实验室数目、分析人员名字或样品类型就是绝对变量的例子.
例如,基体类型变化(在所述方法范围内)的影响可从重复的多基体研究中所进行的回收率研究中估计.
然后,方差分析就能提供所观察到的分析回收的基体内和基体间方差分量.
基体间方差分量能够提供与基体变化有关的标准不确定度.
7.
12基于其他结果或数据的评估7.
12.
1通常可能使用任何现有的关于所考虑量的不确定度的相关信息来评估某些标准不确定度.
下面建议一些信息来源.
7.
12.
2U水平测试(PT)项目U.
实验室参加水平测试的结果可以用于检查已评估的不确定度,因为该不确定度应该与该实验室参加多个回合的水平测试的结果的分布兼容.
特别是在下述特殊情况下:·当水平测试项目所用样品的组成覆盖日常分析的全部范围·当每个回合的所赋值可追溯到适当的参考值,以及·所赋值的不确定度比观察到的结果分布小此时,在重复回合中报告值和所赋值之差的分散较好地为评估在水平测试项目范围内测量过程所产生的测量不确定度提供了基础.
例如,对于具有类似材料和被分析物水平的含量测试项目,结果之差的标准偏差给出了标准不确定度.
当然,对于可溯源的所赋值的CNAS-GL06:2006第29页共148页2006年06月01日发布2006年07月01日实施系统偏差和不确定度的其他来源(如7.
6.
1节中所指的那些)也必须加以考虑.
7.
12.
3U质量保证(QA)数据U:如前所述,必须保证达到标准操作程序中所制定的质量准则.
并且对质量保证样品的测量显示该准则继续得到满足.
在质量保证核查中使用标准物质时,7.
5节中给出了如何使用该数据来评估不确定度.
如果使用了任何其他稳定的物质,质量保证数据提供了一个中间精密度的估计值(7.
7.
2节).
质量保证数据也构成了不确定度所引用数值的持续核查.
很显然,由随机影响产生的合成不确定度不可能小于质量保证测量的标准偏差.
7.
12.
4U供应商的信息U:对于许多不确定度来源,校准证书或供应商的目录可以提供信息.
例如:容量玻璃器皿在使用前,其允差可由生产厂商的产品目录上取得,或从针对特定项目的校准证书上取得.
7.
13根据理论原理建立模型7.
13.
1很多情况下,得到公认的物理理论可以提供结果影响量的良好模型.
例如:温度对体积和密度的影响已了解的很清楚.
在这种情况下,可以使用第8节的不确定度传播方法从关系式评估或计算不确定度.
7.
13.
2在其他情况下,可能有必要将近似的理论模型跟实验数据一起使用.
例如,当分析测量取决于需计时的衍生反应时,可能有必要评估与计时有关的不确定度.
可简单地通过变化所用的时间来达到.
然而,可通过对在所测量浓度附近的衍生动力学的简要实验研究来建立近似的速率模型,并且在给定的时间从所预测的变化速率来评估不确定度,这样做更好.
7.
14基于判断的评估7.
14.
1不确定度的评估既不是日常工作,也不是纯数学推导.
它依赖于对被测量本质和所采用的测量方法和程序的详细了解.
对于作为测量结果所引用的不确定度,其质量和使用最终依赖于对其数值赋值有贡献的那些因素的理解、严格分析和完整性.
7.
14.
2很多数据的分布可以理解为较少观察到其在边界的情况,而较多观察到在中间的情况.
对于这些分布及其相关标准偏差的定量可通过重复测量得到.
7.
14.
3然而,不能重复测量或不能提供特定不确定度分量的有意义测量时,可能需要对于区间的其他评估.
7.
14.
4在分析化学中,有很多例子,后者占主要的情况,因此需要用判断来评估.
例如:CNAS-GL06:2006第30页共148页2006年06月01日发布2006年07月01日实施·不能对每个单一样品进行回收率和其不确定度的评估.
相反,只能对样品类型作出评估(例如,按基体的类型分组),并且结果适用于所有相似类型的样品.
类似程度其本身是一个未知量,因此该推论(从某一类型的基体到某一特定样品)是与不确定度的一个额外分量有关,而对其通常没有说明.
·由分析程序中的规定所定义的测量模型是用来将被测数量值转换为被测量的值(分析结果).
该模型,如科学上的所有模型一样,本身也有不确定度.
只是假设其按特定模型运作,但永远不能100%肯定.
·使用标准物质是极其鼓励的,但仍有不确定度,不仅其真值有不确定度,分析具体样品时特定标准物质的相关性也有不确定度.
因此,在特定情况下需要判断所声明使用的标准物质与样品性质合理接近的程度.
·当程序对被测量定义不充分时会产生另一种不确定度来源.
比如测定"可氧化的高锰酸盐物质"的情况,无论是分析地上的水或是城市废水,毫无疑问都是不同的.
不仅有氧化等因素,基体组成或干扰等化学影响均可能对该技术规定有影响.
·在分析化学中通常的做法是加入单一物质,如结构接近的类似物或同位素标记化合物,相关的内在物质的回收率或甚至整类的化合物的回收率可据此判断.
很明显,假如分析家准备研究在所有浓度水平,以及任何比例的被测量对加入物和所有"相关"基体的回收率,则其不确定度可通过实验评估.
但通常避免进行该实验,而采用下列判断:*浓度与被测量回收率的相关性*浓度与加入物回收率的相关性*回收率与基体(次)类型的相关性*内在物质和加入物的结合模型的性质7.
14.
5此类判断不是基于直接的试验结果,而是基于主观(人为的)的概率,这是一个与'可信程度'、'直觉概率'和'可信性'[H.
11]同义的术语.
也假定可信程度不是基于仓促的判定,而是周密考虑并成熟的概率判定.
7.
14.
6虽然主观概率因人而异,甚至对于同一个人因时而异,这是公认的,但并不武断,因为他们受常识、专业知识和以往经验和观察的影响.
7.
14.
7这可能是一个短处,但是在实际中不必导致比重复测量更坏的评估.
当实验条件的真实、实际的变化性不可模拟、以及因此导致的结果数据的变化不能反映实际情况时特别适用.
CNAS-GL06:2006第31页共148页2006年06月01日发布2006年07月01日实施7.
14.
8当需要对长期的变化性进行评估,而又没有协同研究数据时,就会产生这种性质的典型问题.
科学家没有选择用主观概率代替实际被测概率(当后者不能得到时),就可能忽略了合成不确定度的重要分量,也就最终不如那些利用主观概率的人客观了.
7.
14.
9评估合成不确定度时,可信程度的两个特征是最基本的:·可信程度应被视为区间值,即提供了类似于经典概率分布的上下边界,·将不确定度的'可信程度'分量合成为合成不确定度,同其它方法得到的标准偏差一样,用同样的计算规则.
7.
15偏差的显著性7.
15.
1对所有的已知并显著的系统影响进行修正,这是ISO指南的总要求;7.
15.
2在判定一个已知的偏差是否可合理忽略不计时,建议使用下列方法:ⅰ)在不考虑相关偏差的情况下评估合成不确定度;ⅱ)将偏差与合成不确定度比较;ⅲ)当偏差与合成不确定度比较不显著时,该偏差可以忽略不计;ⅳ)当偏差与合成不确定度比较显著时,需要采取额外的措施.
合适的措施可能包括:·消除或修正偏差,并适当考虑该修正的不确定度.
·除结果外,还要报告所观察到的偏差及其不确定度注:当已知偏差未能按惯例修正时,该方法应视为经验方法(见7.
8节).
8.
第四步计算合成不确定度8.
1标准不确定度8.
1.
1合成前,所有不确定度分量必须以标准不确定度即标准偏差表示.
这可能涉及到将某些度量分散性的其他方法进行转换.
下面的规则为将一个不确定度分量转换成为标准不确定度提供了某些指导原则.
8.
1.
2当不确定度分量是通过试验方法用重复测量的分散性得出时,可以容易用标准偏差的形式表示.
对于单次测量的不确定度分量,标准不确定度就是所观察的标准偏差;对于平均值的结果,使用平均值的标准差【B.
24】.
8.
1.
3当不确定度的评估是源于以前的结果和数据时,可能已经用标准偏差的形式表示了.
如果给出了带有置信水平的置信区间(用±α表示,并指明p%),则将α值除以与所给出CNAS-GL06:2006第32页共148页2006年06月01日发布2006年07月01日实施的置信水平相应的正态分布百分点的值就得到标准偏差.
例子:规范中规定天平的读数为±0.
2mg,置信水平为95%.
从正态分布的百分点标准表上,95%的置信区间用1.
96σ值进行计算.
使用这个数值得出标准不确定度约为(0.
2/1.
96)≈0.
1.
8.
1.
4如果限值±α给出时没有给定置信水平,有理由认为可能是极限值,通常假定其为矩形分布,标准偏差为3/α(见附录E).
例子:证书给出10mlA级容量瓶为±0.
2ml,则该标准不确定度为ml12.
03/2.
0≈.
8.
1.
5如果±α的限值给出时没有给定置信水平,但是有理由认为不可能是极限值,通常假定为三角形分布,标准偏差为6/α(见附录E).
例子:证书给出10mlA级容量瓶为±0.
2ml,但日常内部检查表明极限值的可能性极少.
则标准不确定度为ml08.
06/2.
0≈.
8.
1.
6当基于判断作出评估时,可能将分量直接评估为标准偏差.
如果不可能,应对现实可能合理存在的最大偏差(剔除简单错误)进行评估.
如果较小值更加可能时,这一估计应按三角形分布处理.
如果没有理由相信小误差比大误差更加可能时,应按矩形分布处理.
8.
1.
7最常用的分布函数的转换因子见附录E.
1.
8.
2合成标准不确定度8.
2.
1在评估了单个的或成组的不确定度分量并将其表示为标准不确定度后,下一步就是要使用下列程序之一计算合成标准不确定度.
8.
2.
2数值y的合成标准不确定度)(yuc和其所依赖的独立参数nxxxK,,21的不确定度之间的总关系式如下:∑∑====niiniiicxyuxucxxyu,12,12221),()(.
.
.
)),((**ISO指南应用更简短的形式)(yui代替),(ixyu.
其中),,(21Kxxy为几个参数K,,21xx的函数,cBiB为灵敏系数,记做iixyc=/,y对xCNAS-GL06:2006第33页共148页2006年06月01日发布2006年07月01日实施的偏导.
),(ixyu为由ix不确定度引起的y的不确定度.
每个变量的分量),(ixyu正好是其用标准偏差表达不确定度的平方乘以相关的灵敏系数的平方.
这些灵敏系数反应了y值如何随着参数21,xx等的变化而变化.
注:灵敏系数也可以直接根据实验得出,当无法找到可靠的数学表达式时极为有用.
8.
2.
3变量相关时,关系式更复杂了:∑∑≠==+=kinlkikikiniiijixxuccxucxyu,,,122.
.
.
,),()())((其中),(kixxu是ix和kx之间的协方差,ic和kc是8.
2.
2节所描述和评估的灵敏系数.
协方差与相关系数iKr有关11,)()(),(≤≤=ikikkikirrxuxuxxu其中8.
2.
4无论不确定度是与单个参数有关,还是与组参数有关,或者是与整个方法有关,上述通用程序都适用.
然而,如果不确定度分量与整个程序有关时,通常表示为对最终结果的影响.
在这种情况下,或当某个参数的不确定度直接表示为其对y的影响时,灵敏系数等于1.
0.
例子:结果为122mgl,其标准偏差为11.
4mgl.
在这些条件下,与精密度有关的标准不确定度u(y)为11.
4mgl.
为清楚起见忽略其他因素,则该测量所蕴含的模型为y=(计算的结果)+ε其中ε代表了测量条件下的随机变化影响.
因此ε/y等于1.
08.
2.
5除了上述情形,当灵敏系数等于1外,以及除了下列规则1和规则2中所给的特殊例子外,必须使用需要进行偏导或数值当量的本通用程序.
附录E中给出了由Kragten提出的数字计算方法的细节[H.
12],该方法有效利用了电子表格软件,根据已知测量模型,从输入标准不确定度得出合成标准不确定度.
除最简单的情况外,建议使用本方法和其他适合的计算机方法.
8.
2.
6有时,合成不确定度的表达可以采用更为简单的形式.
合成标准不确定度的两个简单规则如下:CNAS-GL06:2006第34页共148页2006年06月01日发布2006年07月01日实施U规则1U:对于只涉及量的和或差的模型,例如:y=(p+q+r+.
.
.
),合成标准不确定度)(yuc如下:.
.
.
.
.
.
)()(.
.
.
)),((22++=qupuqpyucU规则2U:对于只涉及积或商的模型,例如y=(p*q*r*…)或y=p/(q*r*…),合成标准不确定度)(yuc如下:.
.
.
.
.
.
)()()(22++=qquppuyyuc其中,u(p)/p等是参数表示为相对标准偏差的不确定度.
注:减法的处理原则与加法相同,除法与乘法相同.
8.
2.
7合成不确定度分量时,为方便起见,应将原始的数学模型分解,将其变为只包括上述原则之一所覆盖的形式.
例如:表达式(o+p)/(q+r)应分解成两个部分:(o+p)和(q+r).
每个部分的临时不确定度用规则1计算,然后将这些临时不确定度用规则2合成为合成标准不确定度.
8.
2.
8下述例子说明了上述规则的使用.
例1:y=(p-q+r),其中p=5.
02,q=6.
45,r=9.
04,标准不确定度u(p)=0.
13,u(q)=0.
05,u(r)=0.
22.
则,y=5.
02-6.
45+9.
04=7.
6126.
022.
005.
013.
0)(222=++=yu例2:y=(op/qr),其中o=2.
46,p=4.
32,q=6.
38,r=2.
99,标准不确定度u(o)=0.
02,u(p)=0.
13,u(q)=0.
11,u(r)=0.
07.
则y=(2.
46*4.
32)/(6.
38*2.
99)=0.
56024.
0043.
056.
0)(99.
207.
038.
611.
032.
413.
046.
202.
056.
0)(2222=*=+++*=yuyu8.
2.
9很多情况下,不确定度分量的大小随着分析物水平的不同而变化.
例如,回收率的CNAS-GL06:2006第35页共148页2006年06月01日发布2006年07月01日实施不确定度对于高浓度的物质可能要小些,光谱信号可能会近似按与密度相称的比例随机地变化(恒定变化系数).
在这种情况下,必须考虑合成不确定度随被分析物的浓度变化.
方法包括:z将规定的程序或不确定度评估限制到被分析物浓度的小范围内.
z将不确定估计值以相对标准偏差的形式给出.
z明确计算相关性并对给定的结果重新计算不确定度;附录E4中给出了这些方法的其他信息.
8.
3扩展不确定度8.
3.
1最后一步是将合成标准不确定度和所选的包含因子相乘得到扩展不确定度.
扩展不确定度需要给出一个期望区间,合理地赋予被测量的数值分布的大部分会落在此区间内.
8.
3.
2在选择包含因子k的数值时,需要考虑很多问题,包括:·所需的置信水平;·对基本分布的了解;·对于评估随机影响所用的数值数量的了解(见下面的8.
3.
3节).
8.
3.
3大多数情况下,推荐k为2.
然而,如果合成不确定度是基于较小自由度(大约小于6)的统计观察的话,选择这个k值可能不充分.
此时k的选择取决于有效自由度.
8.
3.
4当合成标准不确定度由某个自由度小于6的分量占决定作用时,推荐将k设成与该分量自由度数值以及置信水平(通常95%)相当的学生t分布的双边数值.
表1中给出了t的主要数值.
例子:称量操作的合成标准不确定度,由标准不确定度分量mgucal01.
0=和5次重复实验的标准偏差mgsobs08.
0=合成.
合成标准不确定度mguc081.
008.
001.
022=+=.
很明显基于5次重复性实验并具有5-1=4自由度的重复性分量sBobsB占主要.
因此k要基于学生t分布.
对于自由度为4,置信水平为95%,查表双边数值t为2.
8;因此k值设为2.
8,扩展不确定度U=2.
8*0.
081=0.
23mg.
8.
3.
5对于使用少量的测量来评估大的随机影响,ISO指南[H.
2]对选择k值的选择方法给出了额外的指导,当评估有几个显著分量的自由度时应参照此指南来执行.
8.
3.
6当所涉及的分布是正态分布时,包含因子2(或根据第8.
3.
3-8.
3.
5节选择的包含因子,置信水平设为95%)给出了大约包含95%分布数值的区间.
在没给出分布的情况下,该区CNAS-GL06:2006第36页共148页2006年06月01日发布2006年07月01日实施间并不意味着是在95%的置信水平下的区间.
表1:95%置信(双边)的学生t分布自由度νt112.
724.
333.
242.
852.
662.
59.
不确定度的报告9.
1总则9.
1.
1报告测量结果所需要的信息取决于其预期的用途.
本指南原则如下:·提供足够的信息以便有新的信息或数据时重新评价结果;·多提供些信息总比少提供信息要好.
9.
1.
2当测量的详细情况,包括如何确定不确定度,主要来自已出版的文件时,必须保证所使用的文件是最新的,并且与所使用的方法一致.
9.
2所需要的信息9.
2.
1测量结果的完整报告应包括,或者引用包括下列信息的文件:·根据实验观察值及输入数据进行测量结果及其不确定度计算的方法描述.
·在计算和不确定度分析中使用的所有修正值和常数的数值和来源.
·所有不确定度分量的清单,包括每一个分量是如何评价的完整文件.
9.
2.
2数据和分析的表达方式应能在必要时容易地重复所有重要步骤并重复计算结果:9.
2.
3当需要详尽的报告,包括中间输入数值时,报告应:·给出每一个输入值的数值及其标准不确定度和获取方法的描述.
·给出结果和输入值之间的关系式及其任何偏导数、协方差或用来说明相关性影响的相关系数;·给出每个输入值的标准不确定度的自由度的评估值.
(评估自由度的方法见ISO指CNAS-GL06:2006第37页共148页2006年06月01日发布2006年07月01日实施南【H.
2】).
注:当函数关系极其复杂或没有明确的函数关系时(例如,可能仅以计算机程序的形式存在),可以用通用术语或引用适当的文献来描述上述关系.
在这种情况下,必须清楚地说明结果值及其不确定度是如何得到的.
9.
2.
4报告日常分析的结果时,仅给出扩展不确定度的数值和k值就可能足够了.
9.
3报告标准不确定度9.
3.
1当不确定度是以标准不确定度uBcB的形式表达时(即一个标准偏差),推荐采用下面的形式:"(结果):x(单位)[和]标准不确定度uBcB(单位)(其中该标准不确定度是国际计量学基本和通用术语词汇表(第二版,ISO1993年)所定义的标准不确定度,相当于一个标准偏差)注:当使用标准不确定度时,不建议使用±符号,因为该符号通常与高置信水平的区间有关.
括号[]中的术语根据需要可以忽略或精简.
例子:总氮含量:3.
52%w/w标准不确定度:0.
07%w/w**标准不确定度相当于一个标准偏差9.
4报告扩展不确定度9.
4.
1除非另有要求,结果x应跟使用包含因子k=2(或按8.
3.
3节所描述,给出k值)计算的扩展不确定度U一起给出.
推荐采用以下方式:"(结果):(x±U)(单位)[其中]报告的不确定度[国际计量学基本和通用术语词汇表(第二版ISO1993年)所定义的扩展不确定度]计算时使用的包含因子为2,[其给出了大约95%的置信水平]"括号[]中的术语可以适当省略或精简.
当然,包含因子应加以调整以反映实际使用的数值.
例子:总氮量:(3.
52±0.
14)%w/w**报告的不确定度是扩展不确定度,使用的包含因子是2,对应的置信水平大约是95%.
CNAS-GL06:2006第38页共148页2006年06月01日发布2006年07月01日实施9.
5结果的数值表示9.
5.
1结果及其不确定度的数值表示中不可给出过多的数字位数.
无论是给出扩展不确定度U,还是标准不确定度u,通常不确定度的有效数字不要多于两位的.
测试结果应根据所给出的不确定度进行适当修约.
9.
6与限值的符合性9.
6.
1被测量(例如:有毒物质的含量)法规通常要求被证明在规定限值内.
本文中的测量不确定度明显地有解释分析结果的含义,特别是:·评价符合性时可能需要考虑分析结果的不确定度;·限值中可能已经给出不确定度的允许量.
在评估中需考虑这两个因素.
下面给出了常规的做法.
9.
6.
2假定限值设定时没有给出不确定度的允许值,对于与上限的符合性情况明显有四种情形(见图2):i)结果高出限值超过一个扩展不确定度;ii)结果高出限值,但不超过一个扩展不确定度;iii)测量结果低于限值,但不超过一个扩展不确定度;iv)结果低于限值超过一个扩展不确定度情形i)通常解释为完全不符合.
情形iv)通常解释为符合.
情形ii)和iii)通常需要根据与数据用户的协议分别考虑.
对于与低限符合性的情况,类似的论点适用.
9.
6.
3当已知或相信设定限值时已考虑到不确定度的允许量时,符合性判断有理由只对给出的允许量进行.
当符合性检查是对照在指定的环境中操作的规定方法时,将产生例外的情况.
在该要求中蕴含着一个假设,即该规定方法的不确定度,或至少是复现性,是足够小以至在实际中可以忽略.
在这种情况下,如果提供了适当的质量控制,通常只对特定结果的数值报告符合性.
这通常会在采用本方法的标准中声明.
CNAS-GL06:2006第39页共148页2006年06月01日发布2006年07月01日实施上控限(i)(ii)(iii)(iv)结果高于限值超过结果高于限值但不结果低于限值但不结果低于限值超一个不确定度超过一个不确定度超过一个不确定度过一个不确定度图2不确定度和符合性限值CNAS-GL06:2006第40页共148页2006年06月01日发布2006年07月01日实施附录A例子介绍简介这些例子阐明了第5至7节所描述的不确定度评估技术如何应用到某些典型的化学分析中.
它们均按照流程图所给出的步骤进行.
识别不确定度来源并在因果图中列出(见附录D).
这有助于避免不确定度来源的双重计算,也有助于将能评估合成效应的分量组合在一起.
例1-6阐明了应用附录E.
2的电子表格方法从已计算出的分量),(ixyu计算合成不确定度.
P*P第8.
2.
2节解释了分量u(y,xBiB)计算的原理.
例1-6的每一个例子均有一个介绍性的摘要.
给出了分析方法的概要、不确定来源表格及其各自的分量、不同分量的图形比较、以及合成不确定度.
例1-3分别阐明了通过量化每一个来源的不确定度来估算不确定度.
每一个均给出了与使用容量玻璃器皿进行体积测量和不同称重方法称量质量有关的不确定度的详细分析.
该细节是为了说明目的,不应作为所需详细程度或所采用的方法的通用建议.
对于许多分析,与这些操作有关的不确定度不是显著的,这样详细的评估将是没必要的.
使用这些操作的典型值并适当考虑所使用的质量和体积的实际值就足够了.
例子A1例子A1是一个为AAS制备HNOB3B中镉的标准溶液的非常简单例子.
其目的是说明如何评估体积测量和称重的基本操作所引入的不确定度分量,以及这些分量如何合成得出总的不确定度.
例子A2例子A2是一个通过标准邻苯二甲酸氢钾(KHP)滴定测量来制备氢氧化钠(NaOH)标准溶液的例子.
它包括了例A1所述的简单体积测量和称量的不确定度评估和对与滴定测量有关的不确定度检查.
例子A3例子A3是例A2的延伸,其增加了用制备好的NaOH滴定HCl.
例子A4CNAS-GL06:2006第41页共148页2006年06月01日发布2006年07月01日实施例子A3说明了按7.
7节所述使用内部确认数据,以及如何使用这些数据来评估许多来源的合成效应所引起的不确定度.
也说明了如何评估与方法偏差有关的不确定度.
例A5例子A5说明了按7.
2-7.
8节所述,使用规定程序测量陶瓷上溶出的重金属含量,如何评估这种按标准或"经验"方法结果的不确定度.
其目的是说明如何在缺乏协同实验数据或稳健性试验结果的情况下,有必要考虑方法定义所允许的参数(例如温度、浸泡时间和酸度)范围所引起的不确定度.
当可获取协同研究数据时,该过程变得相当简单,如下一个例子所示.
例A6例子A6是粗(食用)纤维测定的不确定度评估.
因为只使用标准方法所定义的被分析物,所以该方法是经验的方法.
在这个例子中,协同研究数据、内部质量保证核查以及文献研究数据均是现成的,允许使用7.
6节所描述的方法.
内部研究证实该方法的性能达到以协同研究所预期的结果.
该例子说明了使用内部方法性能核查所支持的协同研究数据如何能大大地降低在此情况下不确定度评估所需要的不同分量的数目.
例A7例子A7详细说明了使用IDMS测量水样中铅含量的不确定度评估.
除了识别可能的不确定度来源并用统计方法量化之外,该例子说明了如何有必要按7.
14节所述的基于判断来评估不确定度分量.
使用判断是ISO指南[H.
2]所描述的B类评估的一个特定例子.
CNAS-GL06:2006第42页共148页2006年06月01日发布2006年07月01日实施例子A1:校准标准溶液的制备概要目的由高纯金属(镉)制备浓度约为11000mgl的校准标准溶液.
测量步骤清洁高纯金属的表面以便除以任何金属氧化物的污染.
然后称量金属并将金属溶于容量瓶的硝酸中.
该步骤的各个阶段见下述流程图.
被测量:][.
.
10001=mglVpmCcd其中:cdC:校准标准溶液的浓度][1mgl1000:从[ml]到[l]的换算系数m:高纯金属的质量[mg]p:以质量分数给出的金属纯度V:校准标准溶液的溶液体积不确定度来源的识别有关的不确定度来源见下页的因果图.
不确定度分量的量化数值及其不确定度见表A1.
1.
合成标准不确定度制备17.
1002mgldC校准标准溶液的合成标准不确定度为19.
0mgl.
对合成标准不确定度的不同分量的大小在图A1.
2中以图表形式列出.
清洁金属表面称量金属溶解并稀释结果图A1.
1镉标准溶液的制备CNAS-GL06:2006第43页共148页2006年06月01日发布2006年07月01日实施表A1.
1数值和不确定度描述数值标准不确定度相对标准不确定度u(x)/xp金属纯度0.
99990.
0000580.
000058m金属质量100.
28mg0.
05mg0.
0005V量瓶体积100.
0ml0.
07ml0.
0007cdc校准标准溶液的浓度17.
1002mgl19.
0mgl0.
0009)()/(),(iiixuxyxyu=的数值取自表A1.
3可读性可读性温度校准复现性V纯度c(Cd)m(tare)m(gross)线性复现性复现性线性校准m校准灵敏度灵敏度Vm纯度c(Cd)00.
20.
40.
60.
81u(y,xB1B)(mgГP1P)图A1.
2在制备镉标准溶液的不确定分量CNAS-GL06:2006第44页共148页2006年06月01日发布2006年07月01日实施例A1:校准标准溶液的制备详细讨论A1.
1引言第一个介绍的例子讨论了从相应的高纯金属制备原子吸收光谱(AAS)所使用的校准标准溶液(本例子中在稀3HNO中约为11000mgl).
尽管该例子没有给出完整的分析测量过程,但校准标准溶液的使用几乎是每一次测量的组成部分,因为现代日常分析测量都是比较测量,需使用参考标准以溯源至SI.
A1.
2步骤1:技术规定该第一步骤的目的是写出被测量的清楚描述.
该技术规定包括校准标准溶液制备的描述以及被测量和其所依赖的参数之间的数学关系.
程序关于如何制备校准标准溶液的具体信息通常在标准操作程序(SOP)中给出.
制备由下列各阶段组成:各步骤是:ⅰ)高纯金属的表面用酸混合物来处理以除去任何金属氧化物污染.
清洁方法由金属制造商提供,并且需要照着去做以便获得证书上所声明的纯度.
ⅱ)分别称量容量瓶(100ml)的重量和容量瓶与净化的金属重量.
使用的天平具有0.
01mg的分清洁金属表面称量金属溶解并稀释结果图A1.
3镉标准溶液的制备CNAS-GL06:2006第45页共148页2006年06月01日发布2006年07月01日实施辨率.
ⅲ)1ml的硝酸(65%m/m)和3ml的去离子水加到量瓶中以溶解镉(约100mg,精确称量).
然后用去离子水充满容量瓶直至刻度,并且倒转容量瓶至少30次以充分混合.
计算本例子中的被测量是校准标准溶液的浓度,取决于高纯度金属(Cd)的称重、纯度以及溶解所用的液体体积.
该浓度由下列公式给出:11000=mglVPmccd其中cdC:校准标准溶液的浓度][1mgl1000:从[ml]到[l]的换算系数m:高纯金属的质量[mg]p:以质量分数给出的金属纯度V:校准标准溶液的溶液体积A1.
3步骤2:识别和分析不确定度来源第二步的目的是列出所有影响被测量数值的各个参数的不确定度来源.
U纯度供应商证书上给出的金属(Cd)纯度是99.
99±0.
01%.
因此P是0.
9999±0.
0001.
这些数值取决于清洁高纯度金属表面的有效性.
如果严格按照制造商给出的步骤,就无需将由于金属表面氧化物污染引起的额外不确定度加到证书所给的数值中.
没有信息表明金属100%溶解了.
因此不得不进行重复的制备实验以检查该因素可否被忽略.
U质量m制备的第二阶段涉及到高纯金属的称重.
要制备100ml11000mgl镉溶液.
镉的相应质量由已扣除皮重的称量给出,得出m=0.
10028g.
制造商的说明书确认扣除皮重称量的三个不确定度来源:重复性、可读性(数字分辨率)以及由于天平校准产生的不确定度分量.
该校准操作有两个潜在的不确定度来源,即天平的灵敏度及其线性.
灵敏度可忽略,因为减量称量是用同一架天平在很窄范围内进行.
注:浮力修正不加以考虑,因为所有的称量结果都是按常规在空气中称量[H.
19].
残余的不确定度很小而不加以考虑.
参见附录G的注1.
U体积VCNAS-GL06:2006第46页共148页2006年06月01日发布2006年07月01日实施容量瓶中的溶液体积主要有三个不确定度来源:·确定容量瓶内部体积时的不确定度·充满容量瓶至刻度的变化·容量瓶和溶液温度与容量瓶体积校准时的温度不同不同的因素及其影响见图A1.
4的因果图(见附录D的说明)A1.
4步骤3:不确定度分量的量化在步骤3中,每一个已识别的潜在来源的不确定度的大小,或者使用以前的实验结果直接测量、评估,或者从理论分析导出.
U纯度在证书上给出的Cd的纯度是0.
9999±0.
0001.
由于不确定度数值的其它信息,故假设是矩形分布.
标准不确定度u(P)的值为0.
0001值除以3(见附录E1.
1).
000058.
030001.
0)(==PuU质量m利用校准证书的数据和制造商关于不确定度评估的建议,对Cd质量有关的不确定度进行估算,结果为0.
05mg.
这种评估考虑了前面已识别的三种分量(A1.
3节).
注:质量不确定度的详细计算可能会非常复杂,当质量不确定度是显著的时候,参考可读性可读性温度校准重复性V纯度c(Cd)m(tare)m(mross)线性重复性重复性线性校准m校准图A1.
4Cd标准溶液制备的不确定度灵敏度灵敏度CNAS-GL06:2006第47页共148页2006年06月01日发布2006年07月01日实施制造商提供的资料是很重要的.
在本例中,为简明起见省略了这些计算.
U体积V该体积有三个主要的影响:校准、重复性和温度影响.
ⅰ)校准:制造商提供的容量瓶在20℃的体积为100ml±0.
1ml,给出的不确定度的数值没有置信水平或分布情况信息,因此假设是必要的.
在这里,计算标准不确定度时是假设三角形分布.
mlml04.
061.
0=注:选择三角形分布是因为在一个有效的生产过程中标定值比极限值可能性更高.
最终分布用三角形分布比矩形分布表示更好.
ⅱ)重复性:由于充满容量瓶的变化引起的不确定度可通过该容量瓶的典型样品的重复性实验来评估.
对典型的100ml容量瓶充满10次并称量的实验,得出标准偏差为0.
02ml.
这可直接用作标准不确定度.
ⅲ)温度:根据制造商提供的信息,该容量瓶已在20℃校准,而实验室的温度在±4℃之间变动.
该影响引起的不确定度可通过估算该温度范围和体积膨胀系数来进行计算.
液体的体积膨胀明显大于容量瓶的体积膨胀,因此只需考虑前者即可.
水的体积膨胀系数为14101.
2°*C,因此产生的体积变化为ml084.
0)101.
24100(4±=***±计算标准不确定度时假设温度变化是矩形分布,即mlml05.
03084.
0=三种分量合成得到体积的标准不确定度u(V)mlVu07.
005.
002.
004.
0)(222=++=A1.
5步骤4:计算合成标准不确定度cdc等于][10001=mglVPmccd中间值及其标准不确定度和相对标准不确定度汇总在表A1.
2.
内容数值)(xuxxu)(金属纯度(P)0.
99990.
0000580.
000058金属质量(m)100.
280.
05mg0.
0005量瓶体积V(ml)100.
00.
07ml0.
0007图A1.
2数值和不确定度CNAS-GL06:2006第48页共148页2006年06月01日发布2006年07月01日实施使用这些数值,得校准标准溶液的浓度为17.
10020.
1009999.
028.
1001000=**=mglccd对于这个简单的乘法表达式,与每一个分量有关的不确定度合成如下:112222229.
00009.
07.
10020009.
0)(0009.
00007.
00005.
0000058.
0)()()()(=*=*==++=++=mglmglccuVVummuPPuccucdcdccdcdc使用附录E给出的电子表格方法导出合成标准不确定度))((cdccu更好,因为即使复杂的表达式也能用它.
已填好的电子表格见表A1.
3.
不同参数的分量大小见图A1.
5.
容量瓶体积的不确定度分量是最大的,称量步骤的不确定度分量类似.
镉纯度的不确定度实际上对总的不确定度没有影响.
将合成标准不确定度乘以包含因子2得到扩展不确定度)(cdcU.
118.
19.
02)(=*=mglmglcUcd)()/(),(iiixuxyxyu=的数值取自表A1.
3Vm纯度c(Cd)00.
20.
40.
60.
81u(y,xB1B)(mgГP1P)图A1.
5在制备镉标准溶液的不确定分量CNAS-GL06:2006第49页共148页2006年06月01日发布2006年07月01日实施表A1.
3不确定度的电子表格计算ABCDE1pmV2数值0.
9999100.
28100.
003不确定度0.
0000580.
050.
0745p0.
99990.
9999580.
99990.
99996m100.
28100.
28100.
33100.
287V100.
0100.
00100.
00100.
0789c(Cd)1002.
699721002.
757881003.
199661001.
9983210),(ixyu0.
058160.
49995-0.
701401122),(,)(ixyuyu0.
745290.
003380.
249950.
491961213u(c(Cd)0.
9将参数的值输入到第二行的C2至E2.
其标准不确定度在下面一行(C3-E3).
电子表格将C2至E2值复制到第二列的B5到B7.
使用这些值后得出的结果(c(Cd))见B9.
C5为C2的p值加上其在C3的不确定度.
使用C5-C7数值得出的计算结果见C9.
D和E列按照类似的步骤.
第10行(C10-E10)的值是行(C9-E9)减去B9之值的差.
第10行(C10-E10)值的平方放在第11行(C11-E11),其平方和的结果值在B11.
B13给出合成标准不确定度,也就是B11的平方根.
CNAS-GL06:2006第50页共148页2006年06月01日发布2006年07月01日实施例A2:氢氧化钠溶液的标定概要目的用标准邻苯二甲酸氢钾(KHP)标定氢氧化钠(NaOH)溶液.
测量步骤干燥并称取滴定标准物KHP.
配制NaOH溶液后,将滴定标准物(KHP)溶解,并用NaOH溶液滴定.
具体的测定步骤见流程图A2.
1被测量TKHPKHPKHPNaOHVMPmc=1000[mol/l]其中,NaOHc:NaOH溶液的浓度[mol/l]1000:由[ml]转化为[l]的换算系数HKPm:滴定标准物KHP的质量[g]KHPP:滴定标准物的纯度,以质量分数表示KHPM:KHP的摩尔质量[g/mol]TV:NaOH溶液的滴定体积[ml]不确定来源的识别:图A2.
2的因果关系图标明了不确定度的有关来源.
不确定度分量的量化:表A2.
1列出了各不确定度分量,图A2.
3以图示方式列出.
0.
10214mol/lNaOH溶液的合成标准不确定度为0.
00010mol/l.
称取KHP制备NaOH滴定结果图A2.
1标定NaOHCNAS-GL06:2006第51页共148页2006年06月01日发布2006年07月01日实施表A2.
1:NaOH标定中的数据和不确定度名称数值x标准不确定度u相对标准不确定度u(x)/xRep复现性1.
00.
00050.
0005KHPmKHP的质量0.
3888g0.
00013g0.
00033KHPPKHP的纯度1.
00.
000290.
00029KHPMKHP的摩尔质量24.
2212g/mol0.
0038g/mol0.
000019TVKHP滴定耗用NaOH的体积18.
64ml0.
013ml0.
0007NaOHcNaOH溶液浓度0.
10214mol/l0.
00010mol/l0.
00097终点温度校准复现性V(T)P(KHP)c(NaOH)m(tare)m(mross)线性m(KHP)线性校准M(KHP)校准图A2.
2建立因果图m(KHP)V(T)终点偏差灵敏度灵敏度V(T)00.
02u(y,xB1B)(mmolГP1P)图A2.
3滴定不确定度分量大小M(KHP)P(KHP)m(KHP)重复性c(NaOH)0.
040.
060.
080.
10.
12CNAS-GL06:2006第52页共148页2006年06月01日发布2006年07月01日实施例A2:氢氧化钠溶液的标定详细讨论A2.
1介绍第二个例子讨论的是氢氧化钠(NaOH)溶液浓度的标定实验.
NaOH由滴定标准物邻苯二甲酸氢钾(KHP)标定.
已知NaOH溶液浓度在0.
1mol/l左右.
滴定的终点由自动滴定装置判断,在该装置中装有测定pH曲线形状的组合pH电极.
滴定标准物KHP的有效成分,即与其分子总数有关的自由质子的数目,使标定的NaOH溶液的浓度可溯源至SI国际单位制.
A2.
2步骤1:技术规定步骤1描述了测量的具体步骤,包括列出测定步骤、被测量的数学计算公式及其所依据的参数.
步骤:NaOH溶液的标定包括以下步骤:各步骤是:ⅰ)按供应商的说明,将标准物KHP干燥.
供应商的说明印制在其提供的产品目录中,并且注明了滴定标准物的纯度及其不确定度.
滴定19ml浓度为0.
1mol/l的NaOH溶液大约需要消耗KHP的量为:g388.
00.
11000191.
02212.
204=***称量应使用最后一位为0.
1mg的天平.
ⅱ)配制0.
1mol/l的NaOH溶液.
为配制1升NaOH溶液,大约需要称取4gNaOH.
然而,由于NaOH溶液的浓度是由标准物KHP来测定,而不是直接计算得到,因此不需要与NaOH的分子量或其质量有关的不确定度来源信息.
ⅲ)将称取的滴定标准物KHP溶解于约50ml去离子水中,再以NaOH溶液滴定.
采用自动滴定装置来滴加NaOH溶液,并记录pH曲线.
从自动滴定装置记录的pH曲线判定滴定终点.
计算:被测量,即NaOH溶液的浓度,取决于KHP的质量、纯度、分子量和滴定终点时消耗的NaOH的KHP称重配制NaOH滴定结果图A2.
4氢氧化钠溶液的标定CNAS-GL06:2006第53页共148页2006年06月01日发布2006年07月01日实施体积TKHPKHPKHPNaOHVMPmc=1000[mol/l]其中,NaOHc:NaOH溶液的浓度[mol/l]1000:由[ml]转化为[l]的换算系数KHPm:滴定标准物KHP的质量[g]KHPP:滴定标准物KHP的纯度,以质量分数表示KHPM:KHP的摩尔质量[g/mol]TV:NaOH溶液滴定消耗的体积[ml]A2.
3步骤2:不确定度来源的确定和分析本步骤的目的是确定各主要不确定度来源,了解其对被测量及其不确定度的影响.
本步骤是分析测定的不确定度评估中最困难的,因为一方面有些不确定度来源可能被忽略,另一方面有些不确定度来源可能会被重复计算.
绘制因果图(附录D)是防止这类问题发生的一个可行的方法.
制作因果图的第一步就是先画出被测量计算公式中的四个参数.
然后,分析测定方法的每一步骤,再沿主要影响因素将其它进一步的影响量添加在图中.
对每一个分支干均进行同样的分析,直到影响因素变得微不足道为止,将所有不可忽略的影响均标注在每一个支干上.
U质量UmBKHPBV(T)P(KHP)c(NaOH)M(KHP)图A2.
5建立因果图的第一步m(KHP)CNAS-GL06:2006第54页共148页2006年06月01日发布2006年07月01日实施大约称取388mgKHP来标定NaOH溶液.
称重为减量称量.
因此在因果图上应画出净重称量(mBtareB)和总重称量(mBgrossB)两条支干.
每一次称重都会有随机变化和天平校准带来的不确定度.
天平校准本身有两个可能的不确定度来源:灵敏度和校准函数的线性.
如果称量是用同一台天平且称量范围很小,则灵敏度带来的不确定度可忽略不计.
所有不确定度来源均标注在因果图上(见图A2.
6).
U纯度PUBUKHPUB供应商目录中标注的KHP的纯度介于99.
95%至100.
05%之间.
因此PBKHPB等于0005.
00000.
1±.
如果干燥过程完全按供应商的规定进行,则无其它不确定度来源.
U摩尔质量MUBUKHPUB邻苯二甲酸氢钾(KHP)的传统分子式为CB8BHB5BOB4BK.
该分子的摩尔质量的不确定度可以通过合成各组成元素原子量的不确定度得到.
IUPAC每两年在《纯粹和应用化学杂志》上发表一次包括不确定度评估值的原子量表.
摩尔质量可以直接由该表计算得到;为求简洁,因果图(图A2.
7)省略了各个原子的质量.
U体积VUBUTUB滴定过程借助于20ml的活塞滴定管.
正如前面例子中充满容量瓶一样,NaOH溶液从活塞滴定管滴定的体积有三个同样的不确定度来源.
这三个来源是滴定体积的重复性、体积校准时的不确定度、以及由实验室温度与活塞滴定管校准时温度不一致而带来的不确定度.
此外,终点检测过程也有影响,有两个不确定来源.
1.
终点检测的重复性,它独立于滴定体积的重复性.
重复性V(T)P(KHP)c(NaOH)M(KHP)图A2.
6增加称量步骤不确定度来源的因果图m(KHP)校准校准线性重复性线性m(tare)m(gross)灵敏度灵敏度CNAS-GL06:2006第55页共148页2006年06月01日发布2006年07月01日实施2.
由于滴定过程中吸入二氧化碳及由滴定曲线计算终点的不准确,滴定终点与等当点之间可能存在的系统误差.
以上各项均标明在图A2.
7因果图中.
A2.
4步骤3:不确定度分量的定量步骤2确定的各不确定度来源在步骤3中进行量化,并转化为标准不确定度.
通常,各类实验都至少包含了活塞滴管滴定体积的重复性和称量操作的重复性.
因此,将各重复性分量合并为总试验的一个分量,并且利用方法确认的数值将其量化是合理的,由此导致对因果图的修订,见图A2.
8.
方法确认表明滴定实验的重复性为0.
05%.
该值可直接用于合成不确定度的计算.
U质量UmBKHPBV(T)P(KHP)c(NaOH)M(KHP)图A2.
8因果图(将重复性合并)m(KHP)校准校准线性线性m(tare)m(gross)校准湿度偏差终点重复性V(T)终点m(KHP)灵敏度灵敏度图A2.
7因果图(所有来源)灵敏度灵敏度重复性V(T)P(KHP)c(NaOH)M(KHP)m(KHP)校准校准线性重复性线性m(tare)m(gross)重复性校准温度重复性偏差CNAS-GL06:2006第56页共148页2006年06月01日发布2006年07月01日实施相关称量有:容器和KHP:60.
5450g(观测)容器和减量的KHP:60.
1562g(观测)KHP:0.
3888g(计算)由于引入了前面已经确定的合成重复性,因此没有必要考虑称量的重复性.
天平量程范围内的系统偏移将被抵消.
因此,不确定度仅限于天平的线性不确定度.
线性:天平计量证书标明其线性为15.
0±mg.
该数值是托盘上的实际重量与天平读数的最大差值.
天平制造商自身的不确定度评价建议采用矩形分布将线性分量转化为标准不确定度.
因此,天平的线性分量为mgmg09.
0315.
0=上述分量必须计算二次,一次作为空盘,另一次为毛重,因为每一次称重均为独立的观测结果,两者的线性影响间是不相关的.
由此得到质量mBKHPB的标准不确定度u(mBKHPB)数值为:mgmuKHP13.
0)09.
0(2)(2=*=注1:由于称量均是按常规在空气中进行的,因此不考虑浮力修正[H.
19].
其它不确定度分量太小,不予考虑.
见附录G的注1.
注2:称量滴定标准物时还存在其它问题.
当标准物的温度与天平的温度即使只差1℃,也会产生与重复性分量数量级相当的漂移.
滴定标准物已完全干燥,但称量的环境湿度在约50%的相对湿度时,也会吸收一些湿气.
U纯度UPBKHPBKHPP为1.
0000±0.
0005.
供应商在目录中没有给出不确定度的进一步的信息,因此可将该不确定度视为矩形分布,标准不确定度00029.
03/0005.
0)(==KHPPu.
U摩尔质量UMBKHPB从IUPAC最新版的原子量表中查得的KHP(CB8BHB5BOB4BK)中各元素的原子量和不确定度:元素原子量不确定度标准不确定度C12.
01070008.
0±0.
00046H1.
00794±0.
000070.
000040O15.
9994±0.
00030.
00017CNAS-GL06:2006第57页共148页2006年06月01日发布2006年07月01日实施K39.
0983±0.
00010.
000058对于每一个元素来说,标准不确定度是将IUPAC所列不确定度作为矩形分布的极差计算得到的.
因此相应的标准不确定度等于查得数值除以3.
各元素对摩尔质量的贡献及其不确定度分量为:计算式结果标准不确定度8C0107.
128*96.
08560.
00375H00794.
15*5.
03970.
000204O9994.
154*63.
99760.
00068K0983.
391*39.
09830.
000058上表各数值的不确定度是由前表各元素的标准不确定度数值乘以原子数计算得到的.
KHP的摩尔质量为:molgMKHP/2212.
2040986.
399976.
630397.
50856.
96=+++=上式为各独立数值之和,因此标准不确定度)(KHPMu就等于各不确定度分量平方和的平方根:molgMuKHP/0038.
0000058.
000068.
00002.
00037.
0)(2222=+++=注:由于每个元素对MBUKHPUB的贡献仅是其原子量与原子个数的乘积,那么按照不确定度分量合成的通用规则可以预见每个元素的不确定度可从单个原子分量平方之和来计算,也就是,如对于碳元素,即001.
000037.
08)(2=*=cMu.
然而,请记住该规则只适用于独立的分量,也就是不同测定值的分量.
对于本例,总量是通过单一值乘8得到的.
注意各元素的不确定度分量是独立的,因此可以用常规方式合成.
U体积VUBUTUB1.
滴定体积的重复性:如前所述,该重复性已通过实验合成重复性考虑了.
2.
校准:制造商已给定了滴定体积的准确性范围为±(数值).
对于20ml活塞滴定管,典型数值为ml03.
0±.
假定为三角形分布,标准不确定度为ml012.
06/03.
0=.
注:如果有理由认为出现在中心区的机率大于极值附近时,ISO指南(F.
2.
3.
3)建议采用三角形分布.
例子A1和A2中的玻璃容器均假定为三角形分布(见例子A1中体积不确定度的讨论).
3.
温度:由于对温度缺乏控制而产生的不确定度按前例方式计算,但这一次假定温度的波动范围CNAS-GL06:2006第58页共148页2006年06月01日发布2006年07月01日实施为3±℃(置信水平为95%).
同样用水的膨胀系数4101.
2*℃1得到ml006.
096.
13101.
2194=***因此因温度控制不充分而产生的标准不确定度为0.
006ml.
注:当处理由于环境因素,如温度控制不充分而产生的不确定度时,考虑不同中间值的影响因素之间的相关性是很重要的.
本例中,溶液温度的主要影响是考虑不同溶剂不同的热效应,也就是说溶液与周围温度并不平衡.
因此,本例中,在STP(标准温度和压力)下温度对每种溶液浓度的影响是不相关的,因此作为独立不确定度分量处理.
4.
终点检测误差:滴定是在氩气层下进行的以避免滴定液吸收COB2B带来的误差.
这样的做法就是尽量避免任何偏差,以免修正.
由于是强碱滴定强酸,没有迹象表明由从pH曲线形状判定终点会与等当点不一致.
所以终点判定偏差及其不确定度可以忽略.
VBTB求得为18.
64ml,合并各不确定度分量得到体积VBTB的不确定度u(VBTB).
mlVuT013.
0006.
0012.
0)(22=+=表A2.
2滴定中的数值与不确定度说明数值x标准不确定度u(x)相对标准不确定度u(x)/xrep重复性1.
00.
00050.
0005mBKHPBKHP的重量0.
3888g0.
00013g0.
00033PBKHPBKHP的纯度1.
00.
000290.
00029MBKHPBKHP的摩尔质量204.
2212g/mol0.
0038g/mol0.
000019VBTB滴定KHP用去NaOH的体积18.
64ml0.
013ml0.
0007A2.
5步骤4:合成标准不确定度的计算CBNaOHB由下式计算获得TKHPKHPKHPNaOHVMPmc=1000[mol/l]表A2.
2列出了上述各参数的数值、标准不确定度和相对标准不确定度.
代入上述数值后,得到:10214.
064.
182212.
2040.
13888.
01000=***=NaOHcmol/lCNAS-GL06:2006第59页共148页2006年06月01日发布2006年07月01日实施对于乘法表示式(如上式),按下式使用标准不确定度:lmolccuNaOHNaOH/00010.
000097.
0)(=*=lmolcNaOHcNaOHuccNaOHcNaOHucVVuMMuPPummureprepuccuTTKHPKHPKHPKHPKHPKHPNaOHNaOHc00010.
000097.
0)(00097.
000070.
02000019.
0200029.
0200033.
020005.
02)()()()()()()(22222=*==++++=++++=为简化上式合成标准不确定度的运算,引用了电子表格(见附录E.
2).
表A2.
3为填入适当数值的的电子表格,并加注了解释.
建议检查不同参数的影响大小.
各参数的影响大小可以很直观地用直方图表示.
图A2.
9显示了从表A2.
3计算得到的),(jxyu的大小.
滴定体积TV的不确定度分量是最大的,其次是重复性.
称量过程和滴定标准物的纯度处于同一数量级,而摩尔质量的不确定度却几乎小了一个数量级.
A2.
6步骤5:重新评估显著性不确定度分量V(T)不确定度分量是最大的.
滴定KHP消耗NaOH的体积V(T)受四种量的影响:滴定所消耗体积的重复性、活塞滴定管的校准、滴定管滴定时的温度与滴定管校准时的温度之间差异,以及终点判定的重复性.
对比各分量的大小,校准是最大的.
所以该分量必须研究透彻.
V(T)00.
02u(y,xB1B)(mmolГP1P)图A2.
9标定NaOH的不确定度分量大小M(KHP)P(KHP)m(KHP)重复性c(NaOH)0.
040.
060.
080.
10.
12CNAS-GL06:2006第60页共148页2006年06月01日发布2006年07月01日实施V(T)校准的标准不确定度是由供应商假定为三角形分布计算得到的数据.
分布形状选择的影响见表A2.
4.
根据ISO指南4.
3.
9注1:"对于正态分布,期望值为μ,标准偏差为α,区间μ±3α覆盖了99.
97%的分布.
因此,如果上下限aB+B和aB-B规定了99.
73%界限,而不是100%的界限,则jX可以假定大致为正态分布,而不是对jX(在区间内)所知无几,那么,9/)(22α=jxu.
相对地,半宽度为a的对称矩形分布的方差为3/2a….
.
而半宽度为a的对称三角形分布的方差为6/2a.
相对于这三种分布的假定的差别,这三种分布的方差却处于同一数量级是非常令人惊讶的.
"因此,影响量的分布函数的选择对合成标准不确定度(u())NaOHc数值的影响并不明显,所以将之假定为三角形分布有充分的理由.
扩展不确定度)(NaOHcU可由合成标准不确定度乘以包含因子2后得到.
lmolcUNaOH/0002.
0200010.
0)(=*=所以,NaOH溶液的浓度为(0.
1021±0.
0002)mol/l.
表A2.
3:滴定不确定度的电子表格计算ABCDEFG1Repm(KHP)P(KHP)M(HP)V(T)2数值1.
00.
38881.
0204.
221218.
643不确定度0.
00050.
000130.
000290.
00380.
01345rep1.
01.
00051.
01.
01.
01.
06m(KHP)0.
38880.
38880.
388930.
38880.
38880.
38887P(KHP)1.
01.
01.
01.
000291.
01.
08M(KHP)204.
2212204.
2212204.
2212204.
2212204.
2250204.
22129V(T)18.
6418.
6418.
6418.
6418.
6418.
6531011c(NaOH)0.
1021360.
1021870.
1021700.
1021660.
1021340.
10206512),(ixyu0.
0000510.
0000340.
000030-0.
000002-0.
0000711322),(,)(ixyuyu9.
72E-92.
62E-91.
16E-99E-104E-125.
041E-91415))((NaOHcu0.
000099CNAS-GL06:2006第61页共148页2006年06月01日发布2006年07月01日实施各参数的数值列于第二行C2-G2.
它们的标准不确定度列于下一行C3-G3.
将C2-G2的数据用电子表格程序复制到列B5-B9.
运用上述数据计算的结果(c(NaOH))列于B11.
C5是C2加上它的不确定度C3得到的重复性数值.
由C5-C9计算得到的结果列于C11.
列D和G重复以上步骤得到.
第12行(C12-G12)的数值是行(C11-G11)减去B11的数值得到的.
第13行(C13-G13)是第12行(C12-G12)的平方,相加后得到的数值列入B13.
B15是合成标准不确定度,它等于B13的平方根.
表A2.
4:不同分布假定的影响分布因子u(V(T;cal))(ml)u(V(T))(ml)))(NaOHccu(mol/l)矩形30.
0170.
0190.
00011三角形60.
0120.
0150.
00009正态P(注1)P90.
0100.
0130.
000085注1:因子9来源于ISO导则4.
3.
9注1的因子3(详见48页).
CNAS-GL06:2006第62页共148页2006年06月01日发布2006年07月01日实施例A3:酸碱滴定概要目标以已知浓度的氢氧化钠(NaOH)溶液标定盐酸(HCl)溶液.
测定步骤以邻苯二甲酸氢钾(KHP)标定氢氧化钠(NaOH)溶液,再以氢氧化钠(NaOH)溶液标定盐酸(HCl)的浓度.
其过程如图A3.
1所示.
)()/(),(iiixuxyxyu=的数值取自表A3.
3图A3.
1滴定过程被测量:HClKHPT1T2KHPKHPHClVMVVPm1000c=[moll-1]其中,上式各代号已在表A3.
1中列出.
1000是由ml转化为升的换算系数.
不确定度来源的确定:各相关不确定度来源如图A3.
2所示.
不确定度分量的定量:最终不确定度评估值为0.
00016mol/l.
表A3.
1列出了各数值及其不确定度;图A3.
3以直方图表示了各数值大小.
称取KHP以NaOH滴定KHP图A3.
1滴定过程准确移取以NaOH滴定HCL结果V(HCI)0u(y,xB1B)(mmolГP1P)u(y,xB1B)=(y/xB1B)·u(xB1B)图A3.
3酸碱滴定不确定度分量M(KHP)V(T1)V(T2)P(KHP)m(KHP)复现性с(HCI)0.
050.
10.
150.
2CNAS-GL06:2006第63页共148页2006年06月01日发布2006年07月01日实施表A3.
1酸碱滴定数据及其不确定度名称数值x标准不确定度u(x)相对标准不确定度u(x)/xrep重复性10.
0010.
001KHPmKHP的质量0.
3888g0.
00012g0.
00033KHPPKHP的纯度1.
00.
000290.
000292TV滴定HCl消耗NaOH的体积14.
89ml0.
014ml0.
000941TV滴定KHP消耗NaOH的体积18.
64ml0.
015ml0.
00080KHPMKHP的摩尔质量204.
2212g/mol0.
0038g/mol0.
000019HClV用NaOH滴定移取的HCl的体积15ml0.
011ml0.
00073HClCHCl溶液的浓度0.
10139mol/l0.
00018mol/l0.
0018湿度校准重复性V(T1)P(KHP)c(HCl)m(tare)m(gross)线性线性校准M(KHP)校准图A3.
2酸碱滴定的因果图m(KHP)m(KHP)终点偏差灵敏度灵敏度V(T2)终点偏差湿度校准V(T2)终点V(T2)V(T1)终点V(T1)V(HCI)湿度校准V(HCI)同一部天平CNAS-GL06:2006第64页共148页2006年06月01日发布2006年07月01日实施例A3:酸碱滴定详细讨论A3.
1介绍本例探讨了标定盐酸(HCl)溶液浓度的一系列实验.
另外,也对滴定的技术进行了多方面的探讨.
HCl用刚由邻苯二甲酸氢钾(KHP)标定的氢氧化钠(NaOH)溶液标定.
与例A2一样,假定HCl的浓度在0.
1mol/l这个数量级,滴定终点用自动滴定装置通过pH曲线的形状判断.
上述分析使测量不确定度以SI单位测量术语给出.
A3.
2步骤1:技术规定步骤1详细叙述了测量步骤,包括罗列了一系列的测量步骤和被测量的数学表达式.
步骤测定HCl溶液浓度包括以下各个阶段(可参见图A3.
4)1)干燥滴定标准物邻苯二甲酸氢钾(KHP),以确保其纯度符合其供应商提供的证书上所标数值.
称取大约0.
388g干燥的基准KHP以标定19mlNaOH.
2)将滴定标准物KHP溶解于约50ml的去离子水中,以NaOH滴定.
滴定装置自动地滴加NaOH,同时绘出pH曲线.
通过记录的pH曲线形状确定终点.
3)用移液管移取15mlHCl溶液.
用去离子水稀释至约50ml于滴定瓶中.
4)用同一台自动滴定装置测定HCl溶液的浓度.
称取KHP以NaOH滴定KHP图A3.
4HCI溶液浓度的测定准确移取HCI以NaOH滴定HCL结果CNAS-GL06:2006第65页共148页2006年06月01日发布2006年07月01日实施计算:被测量是HCl溶液的浓度CBHClB.
它取决于KHP的质量、纯度、分子量、两次滴定终点时消耗NaOH的体积和HCl的移取量:HClKHPTTKHPKHPHClVMVVPmc=121000[mollP-1P]其中CBHClB:HCl溶液的浓度[mol/l]1000::由ml转化为l的换算系数mBKHPB::称取的KHP质量[g]PBKHPB:KHP以质量分数表示的纯度VBT2:B滴定HCl所用NaOH溶液的体积VBT1:B滴定KHP所用NaOH溶液的体积MBKHPB:KHP的摩尔质量[g/mol]VBHClB:被NaOH滴定的HCl的体积[ml]A3.
3步骤2:确定和分析不确定度来源用直观的因果图(图A3.
5)表示出各不确定来源及其对被测量的影响是分析测量不确定度分量的最好方法.
重复性评估作为整体可以方法确认研究中得到,因此无需分别考虑所有重复性的分量.
因此这些湿度校准重复性V(T1)P(KHP)c(HCI)m(tare)m(gross)线性线性校准M(KHP)校准m(KHP)m(KHP)终点偏差灵敏度灵敏度V(T2)终点偏差湿度校准V(T2)终点V(T2)V(T1)终点V(T1)V(HCI)湿度校准V(HCI)同一部天平图A3.
5最终因果图CNAS-GL06:2006第66页共148页2006年06月01日发布2006年07月01日实施可以归纳为一种分量(见修订后的因果图A3.
5).
对参数VBT2B、VBT1B、MBKHPB、PBKHPB和MBKHPB的影响因素已在前几个例子中有详尽的探讨,因此只有VBHClB影响量在本节中详细讨论.
U体积VUBUHClUB用移液管移取15ml待测HCl溶液.
从移液管排出的HCl溶液的体积与所有容量测量装置一样有三种同样的不确定度来源:1.
排出体积的变化或重复性2.
移液管所标体积的不确定度3.
移液管移取的溶液温度与校准时温度的差异.
A3.
4步骤3:不确定度分量的量化本步骤的目的就是将步骤2分析的各不确定度来源进行定量.
各分支或不同组分的定量在前两个例子中有详细的描述.
因此,本节只给出不同分量的概要.
U重复性方法确认表明测定的重复性为0.
1%(%rsd).
该数值可以直接用来计算与各重复性有关的合成标准不确定度.
U质量UmBKHPB校准/线性:天平制造商给出了±0.
15mg的线性分量.
该数值代表了托盘上被称量的实际重量与从天平所读取的数值的最大差值.
线性分量被假设成矩形分布,换算成标准不确定度为:mg087.
0315.
0=线性分量应重复计算两次,一次是空盘,另一次为毛重,产生的不确定度u(mBKHPB)为:2)087.
0(2)(*=KHPmu=0.
12mg注1:由于对非线性的形式未做任何假设,因此将该分量重复计算了两次.
非线性被相应地看作为对每次称重的系统影响,在称量范围内影响的大小是随机变化的.
注2:因为所有称重均是以常规方式在空气中完成的,因此未考虑浮力修正[H.
19].
余下的不确定度太小,可以忽略不计.
参见附录G中的注1.
UP(KHP)供应商证书上给出的P(HKP)值为100%±0.
05%,其引用的不确定度可考虑为矩形分布,标准不确CNAS-GL06:2006第67页共148页2006年06月01日发布2006年07月01日实施定度为:00029.
030005.
0)(==puKHPUV(T2)ⅰ)校准:制造商提供的数值(±0.
03ml),近似于三角形分布0.
03/6=0.
012mlⅱ)温度:温度变化的范围为±4℃,近似于矩形分布15*2.
1*10P-4P*4/3=0.
007mlⅲ)终点判定偏差:在氩气中滴定可以消除空气中的COB2B造成判定的终点与等当点的偏差.
不确定度可不予考虑.
VBT2B求得为14.
89ml,将两个分量合成为体积VBT2B的不确定度μ(VBT2B):mlVuT014.
0007.
0012.
0)(222=+=U体积VUBUT1UB除温度外,所有分量均与VBT2B相同.
ⅰ)校准:0.
03/6=0.
012mlⅱ)温度:滴定0.
3888gKHP大约消耗NaOH的体积为19ml,因此其不确定度分量为19*2.
1*10P-4P*4/3=0.
009mlⅲ)偏差:可忽略VBT1B求得为18.
64ml,其标准不确定度u(VBT1B):mlVuT015.
0009.
012.
0)(221=+=U摩尔质量MUBKHPBKHP(CB8BHB5BOB4BK)各组成元素的原子量及其不确定度(从最新的IUPAC原子量表查得)为:元素原子量不确定度标准不确定度C12.
0107±0.
00080.
00046H1.
00794±0.
000070.
000040O15.
9994±0.
00030.
00017K39.
0983±0.
00010.
000058对于每个元素来说,其标准不确定度可按IUPAC给出的数值以矩形分布求得.
将所给的数值除以3可得到其标准不确定度.
CNAS-GL06:2006第68页共148页2006年06月01日发布2006年07月01日实施KHP摩尔质量MBKHPB及其不确定度分别为:MBKHPB=8*12.
0107+5*1.
00794+4*15.
9994+39.
0983=204.
2212g/molu(MBKHPB)=2222000058.
0)00017.
04()00004.
05()00046.
08(+*+*+*=0.
0038g/mol注:单个原子的不确定度分量并非独立的.
因此计算原子分量的不确定度时将原子量的标准不确定度乘以原子数.
U体积VUBUHClUBⅰ)校准:制造商给定15ml移液管的不确定度为±0.
02ml,近似为三角形分布0.
02/6=0.
008mlⅱ)温度:实验室温度变化介于±4℃之间.
采用矩形分布,其标准不确定度为:15*2.
1*10P-4P*4/3=0.
007ml合成上述不确定度分量:u(VBHClB)=222007.
0008.
00037.
0++=0.
011ml表A3.
2酸碱滴定数据及其不确定度(两次滴定过程)名称数值x标准不确定度u(x)相对标准不确定u(x)/xrep重复性10.
0010.
001KHPmKHP的质量0.
3888g0.
00012g0.
00031KHPPKHP的纯度1.
00.
000290.
000292TV滴定HCl用去NaOH的体积14.
89ml0.
014ml0.
000941TV滴定KHP用去NaOH的体积18.
64ml0.
015ml0.
00080KHPMKHP的摩尔质量204.
2212g/mol0.
0038g/mol0.
000019HClV滴定NaOH移取HCl的体积15ml0.
011ml0.
00073A3.
5步骤4:计算合成标准不确定度cBHClB由下式计算得到:HClKHPTTKHPKHPHClVMVVPmc=121000注:本例中,重复性评估视为相对影响;因此完整的公式应为CNAS-GL06:2006第69页共148页2006年06月01日发布2006年07月01日实施repVMVVPmcHClKHPTTKHPKHPHCl*=121000先后两次滴定实验的所有中间值及其标准不确定度均列于表A3.
2中.
代入这些数值后110139.
01152212.
20464.
1889.
140.
13888.
01000=******=mollcHCl相应地合成各不确定度分量:lmolcHClcHClucrepuVVuMMuVVuVVuPPummuccuHClHClKHPKHPTTTTKHPKHPKHPKHPHClHCl00018.
00018.
0)(0018.
0001.
000073.
0000019.
000080.
000094.
000029.
000031.
0)()()()()()()()(222222222221122222=*==++++++=++++++=电子表格方法(见附录E)可用于简化上述合成不确定度的计算.
表A3.
3中的电子表格已填入了相应数值,并附有相应的解释.
不同不确定度分量的大小可以用直方图的形式比较.
图A3.
6将表A3.
3中各分量),(jxyu的值用直方图表示.
将合成标准不确定度乘以包含因子2计算扩展不确定度U(cBHClB):U(cBHClB)=0.
00018*2=0.
0004mol/lHCl溶液的浓度为:(0.
1014±0.
0004)mol/lV(HCI)0u(y,xB1B)(mmolГP1P)图A3.
6酸碱滴定的不确定度M(KHP)V(T1)V(T2)P(KHP)m(KHP)重复性с(HCI)0.
050.
10.
150.
2CNAS-GL06:2006第70页共148页2006年06月01日发布2006年07月01日实施表A3.
3酸碱滴定----电子表格计算不确定度ABCDEFGHI1repm(KHP)P(KHP)V(T2)V(T1)M(KHP)V(HCl)2数值1.
00.
38881.
014.
8918.
64204.
2212153不确定度0.
0010.
000120.
000290.
00140.
0150.
00380.
01145rep1.
01.
0011.
01.
01.
01.
01.
01.
06m(KHP)0.
38880.
38880.
388920.
38880.
38880.
38880.
38880.
38887P(KHP)1.
01.
01.
01.
000291.
01.
01.
01.
08V(T2)14.
8914.
8914.
8914.
8914.
90414.
8914.
8914.
899V(T1)18.
6418.
6418.
6418.
6418.
6418.
65518.
6418.
6410M(KHP)204.
2212204.
2212204.
2212204.
2212204.
2212204.
2212204.
2250204.
221211V(HCl)1515151515151515.
0111213c(HCl)0.
1013870.
1014890.
1014180.
1014170.
1014820.
10130601013850.
10131314),(xyu0.
0001010.
0000310.
0000290.
000095-0.
000082-0.
0000019-0.
0000741522),(,)(ixyuyu3.
34E-81.
03E-89.
79E-108.
64E-109.
09E-96.
65E-93.
56E-125.
52E-91617))((HClcu0.
00018各参数的数值列于第二行C2到I2.
它们的标准不确定度列于下一行(C3-I3).
用电子表格将C2-I2的数值复制到第二列从B5到B11.
利用上述数据计算得到的结果(c(HCl))列于B13.
C5显示的是重复性的数值,它等于C2加上其不确定度C3.
将C5-C11的数据计算的结果列于C13.
列D至列I重复类似的过程.
第14行(C14-I14)的数值是行(C13-H13)减去B13的结果.
第15行(C15-I15)是第14行(C14-I14)的平方,其平方和列于B15.
B17给出了合成标准不确定度,等于B15的平方根.
A3.
6滴定例子中的特殊性滴定实验的特殊性在本例第二部分中进行探讨.
研究实验准备或滴定过程中哪些因素的变化会对最终结果产生影响以及其不确定度很有趣的事.
U25℃平均室温的影响对于常规分析,分析化学师很少会对实验室温度对体积的系统影响进行修正.
这种情况涉及到修CNAS-GL06:2006第71页共148页2006年06月01日发布2006年07月01日实施正所引入的不确定度.
体积测量装置是在20℃条件下校准的.
但是很少有实验室配备了温度控制装置来保持在该温度.
为了说明,考虑对25℃的平均室温为进行修正.
应使用修正后的体积来计算最终分析结果,而不是按20℃时校准的体积计算.
按照下式,修正温度对体积的影响:)]20(1['=TVVα其中V':平均温度T时的实际体积V:20℃时的校准体积α:水溶液的膨胀系数[℃P-1P]T:实验室实际观测到的温度[℃]被测量的计算公式应重写为:''1'21000HClTTKHPKHPKHPHClVVVMPmc=加入温度修正项后:()[]()[]()[]*==2012012011000100012''1'2TVTVTVMPmVVVMPPmHClcHClTTKHPKHPKHPHClTTKHPKHKHPααα上式在假设温度T和水溶液的膨胀系数α对于三个体积均是相同的,可以简化为:*=)]20(1[100012TVVVMPmcHCITTKHPKHPKHPHCIα上式给出的HCl结果与20℃时称有差别:1410149.
0)]2025(101.
21[1564.
182236.
20489.
140.
13888.
01000=*******=mollcHCI该数值仍然落在平均温度为20℃时结果的合成标准不确定度给出的范围内,说明结果无显著影响.
由于25℃平均室温的温差变化假设为±4℃,因此,温度的变化并不影响合成标准不确定度的评价.
U肉眼判断滴定终点CNAS-GL06:2006第72页共148页2006年06月01日发布2006年07月01日实施如果用酚酞代替由pH曲线求等当点的自动终点识别装置作终点判断,就会引入误差.
由透明向红/粉红色转变的pH值介于8.
2至9.
8之间,这会额外增加滴定量,相对测定pH值的自动终点识别装置来说引入了误差.
研究显示这会使体积增加0.
05ml,同时肉眼判断的标准不确定度大约为0.
03ml.
额外增加的体积必须在最终的结果中考虑到.
肉眼判断时实际的体积为:ExcessTIndTVVV+=1;1IndTV;1:肉眼判断终点时的体积1TV:等当点时的体积ExcessV:使酚酞变色需要增加的体积上述体积的修正导致被测量计算式的改变:HCIExcessIndTKHPExcessIndTKHPKHPHCIVVVMVVPmc=)()(1000;1;2标准不确定度u(VBT2B)和u(VBT1B)必须用肉眼判断终点的标准不确定度作为终点判断重复性的不确定度分量重新计算,mlVVuVuExcessIndTT034.
003.
0009.
0012.
0004.
0)()(2222;11=+++==mlVVuVuExcessIndTT033.
003.
0007.
0012.
0004.
0)()(2222;22=+++==合成标准不确定度)(HClccu=0.
0003mol/l比以前大得多.
U三次测定求得最终结果两次滴定实验重复了三次后,求得最终结果.
三次测定可减少重复性的分量,减少总不确定度.
正如本例第一部分所述,所有重复实验的变化都合并成一个组分,作为总的实验重复性,见图A3.
5所示的因果关系图.
各不确定度分量通过下列方式计算:U质量UmBKHPB线性:0.
15/mg087.
03=mgmuKHP12.
087.
02)(2=*=U纯度UPBKHPB纯度:00029.
03/005.
0=CNAS-GL06:2006第73页共148页2006年06月01日发布2006年07月01日实施U体积VUBUT2UB计量:ml012.
06/03.
0=温度:ml007.
03/4101.
2154=***mlVuT014.
0007.
0012.
0)(222=+=U重复性三次测定的质量记录显示长期的实验平均标准偏差为0.
001(以RSD表示).
不建议使用三次测定的实际标准偏差,因为它本身包含了52%的不确定度.
三次测定(次独立测定)的标准不确定度由0.
001除以3得到:00058.
03/001.
0Re==p(RSD)U体积UHClV校准:ml008.
06/02.
0=温度:ml007.
03/4101.
2154=***mlVuKHP01.
0007.
0008.
0)(22=+=U摩尔质量MUBUKHPUBU:0038.
0)(=KHPMug/molU体积U1TV校准:ml02.
06/03.
0=温度:ml009.
03/4101.
2194=***mlVuT015.
0009.
0012.
0)(221=+=所有不确定度分量的值均收于表A3.
4.
合成标准不确定度为0.
00016mol/l,由于是三次测定因此是略有下降.
各不确定度分量的直方图图示对比见图A3.
7,可明显看出那个结果的一些理由.
虽然重复性分量大大降低了,体积的不确定分量仍保持原来的数值,限制了进一步的改善.
V(HCI)0u(y,xB1B)(mmolГP1P)图A3.
7重复实验酸碱滴定的值和不确定度M(KHP)V(T1)V(T2)P(KHP)m(KHP)复现性с(HCI)0.
050.
10.
150.
2重复实验单次实验CNAS-GL06:2006第74页共148页2006年06月01日发布2006年07月01日实施表A3.
4重复酸碱滴定的数值和不确定度名称数值x标准不确定度u(x)相对标准不确定u(x)/xrep重复性1.
00.
000580.
00058KHPmKHP的质量0.
3888g0.
00013g0.
00033KHPPKHP的纯度1.
00.
000290.
000292TV滴定HCl用去NaOH的体积14.
90ml0.
014ml0.
000941TV滴定KHP用去NaOH的体积18.
65ml0.
015ml0.
0008KHPMKHP的摩尔质量204.
2212g/mol0.
0038g/mol0.
000019HClV滴定NaOH移取出HCl的体积15ml0.
01ml0.
00067CNAS-GL06:2006第75页共148页2006年06月01日发布2006年07月01日实施例A4实验室内部确认研究的不确定度评估面包中有机磷农药的测定概要目的使用萃取和GC方法测定面包中残留的有机磷农药的含量.
测定程序测定面包中残留的有机磷农药所需的步骤见图A4.
1.
被测量16hom10Re=mgkgFmcIVCIPsamplerefoprefopop其中:opp:样品中农药的含量水平][1mgkgopI:样品萃取液的峰响应值refC:参考标准的质量浓度][1gmlμopV:萃取样品的最终体积[ml]610:从][1gg到[1mgkg]的换算因子refI:参考标准的峰响应值Rec:回收率samplem:待测子样品的质量[g]homF:样品非均匀性校正因子确定不确定度来源相关不确定度来源见图A4.
2的因果图.
不确定度分量的量化基于实验室内部确认数据,三个主要的分量见表A4.
1,直方图表示见图A4.
3(数值取自表A4.
5).
混合均匀萃取图A4.
1有机磷农药分析过滤稀释定容GC测定制备校准标准GC校准结果CNAS-GL06:2006第76页共148页2006年06月01日发布2006年07月01日实施表A4.
1:农药分析的不确定度描述数值x标准不确定度u(x)相对标准不确定度u(x)/x评述重复性(1)1.
00.
270.
27基于不同类型样品的平行试验偏差(回收率)(2)0.
90.
0430.
048加料样品其它来源(3)(均匀性)1.
00.
20.
2基于假设模型的评估u(PBopB)/PBopB----0.
34相对标准不确定度F(hom)P(op)图A4.
2农药分析的不确定度来源重复性m(样品)V(op)稀释V(ref)m(ref)l(ref)l(op)校准t(op)V(ref)m(ref)稀释校准温度校准校准线性c(ref)纯度(ref)V(op)V(op)校准温度回收率l(ref)校准m(样品)m(gross)校准线性m(tare)校准线性均匀性偏差重复性P(op)00.
10.
20.
30.
4u(y,xB1B)(mgГP1P)u(y,xB1B)=(y/xB1B)·u(xB1B)的数值取自表A4.
5图A4.
3农药分析的不确定度CNAS-GL06:2006第77页共148页2006年06月01日发布2006年07月01日实施混合均匀萃取图A4.
4有机磷农药分析过滤稀释定容GC测定制备校准标准GC校准结果例A4面包中有机磷农药的测定详细讨论A4.
1介绍这个例子说明了用实验室内部确认的数据计算测量不确定度的方法.
本测量的目的是测定面包中残余有机磷农药的含量.
通过测定加料样品,实验室确认计划和实验建立起溯源性.
假设对样品中所加的料和被分析物的测量响应的不同而导致的不确定度相对于结果的总不确定度是小的.
A4.
2步骤1:技术规定有关被测量更详细分析方法的技术规定最好是综合描述分析方法的不同的步骤,并提供被测量的计算公式.
程序:测定程序在图A4.
4中以图的方式说明.
各步骤如下:ⅰ)均匀性:将完整的样品切成小碎片(约2cm),从中随机选择15个小碎片,并把该子样品混合均匀.
当观察到极度不均匀性时,在混合前按比例取样.
ⅱ)称量分析用的子样品,给出samplemⅲ)萃取:用有机溶剂定量萃取被分析物,转移并通过硫酸钠的柱子进行脱水,用Kuderna-Danish装置浓缩萃取液.
ⅳ)液-液萃取:ⅴ)乙腈/己烷分配液,用己烷洗乙腈萃取液,通过硫酸钠柱干燥已烷层.
ⅵ)浓缩洗过的萃取液,用气体把萃取液吹至近干.
ⅶ)稀释至标准体积opV(约2ml),用10ml刻度管.
ⅷ)测定:注射5μl样品萃取液至GC中,得到峰响应值opI.
ⅸ)制备约1.
5mlgμ标准溶液(实际质量浓度为refC).
ⅹ)用制备的标准溶液作GC校准.
注射5l标准溶液到GC中,得到参考标准峰响应值refI.
计算:CNAS-GL06:2006第78页共148页2006年06月01日发布2006年07月01日实施最终样品萃取液的质量浓度opc为:1=gmlIIccrefoprefopμ散样中的农药含量PBopB估计值为:1610Re=mgkgmcVcPsampleopopop或将opC带入得:1610Re=mgkgmcIVcIPsamplerefoprefopop其中:opp:样品中农药的含量水平[1mgkg]opI:样品萃取液的峰响应值refC:参考标准的质量浓度[1gmlμ]opV:萃取液的最终体积[ml]610:从[1gg]到[1mgkg]的换算因子refI:参考标准的峰响应值Rec:回收率samplem:待测子样品的质量[g]范围:此分析方法适用于小范围的化学性质相近,含量介于0.
01至2mg1kg之间的各类的面包基体中的农药.
A4.
3步骤2识别和分析不确定度来源对这样一个复杂的分析程序,识别所有的不确定度来源最好方法是画一个因果图.
被测量计算公式中的参数在图作为主要分支.
考虑到分析程序(A4.
2)的每一步,进一步将影响因素添加到该图上,直到所有重要贡献因素均予充分考虑.
CNAS-GL06:2006第79页共148页2006年06月01日发布2006年07月01日实施样品的非均匀性不是原始被测量公式中的一个参数,但是它在分析程序中对分析结果有很大的影响.
因此在因果图中加上了一个新的分支,(hom)F,代表样品的非均匀性(图A4.
5).
最后,样品非均匀性引入的不确定度分量应该包括进被测量的计算公式.
为了明确地表明非均匀性引起的不确定度的影响,被测量计算公式应写成下式是有益的:][1016hom=mgkgmRIVcIFPsampleecrefoprefopop其中homF是校正因子,在原先计算公式中设为1.
这就明确了校正因子的不确定度必须包括在总的不确定度的评价中.
这个最终的公式表明了不确定度是怎样应用的.
注:校正因子:这个方法非常概括,可能对突出那些隐藏着的假设很有用.
原则上,每次测量都与这些通常设为1的校正因子联系在一起.
例如,opc的不确定度可以被表示为opc的标准不确定度,或是表示为校正因子不确定度的标准不确定度.
对于后者,该数值等同于以相对标准偏差表示的opc的不确定度.
A4.
4步骤3:不确定度分量的量化根据第7.
7节,不同的不确定度分量的量化可以利用实验室内部开发和确认研究中的数据.
·分析过程随机变化总体的现有的最佳估计值·总体偏差(回收率)及其不确定度可能的最佳估计值F(hom)P(op)图A4.
5将样品非均匀性作为一个主要分支后的因果图校准l(op)V(ref)m(ref)稀释校准温度校准校准线性c(ref)纯度(ref)V(op)V(op)校准温度回收率I(ref)校准m(样品)m(gross)校准线性m(tare)校准线性精密度精密度精密度精密度精密度精密度精密度灵敏度灵敏度精密度CNAS-GL06:2006第80页共148页2006年06月01日发布2006年07月01日实施·总体性能研究不能全面考虑的其他影响因素有关的不确定度的量化为使这些输入数据的关系和范围更加明确,有必要对因果图做些重新安排(图A4.
6).
注:通常,测量用的样品按小批量来管理,每一批量包括一个校准样,控制偏差的回收率检查样品,以及检查精密度的随机平行样.
假如这些检查表明显著地偏离确认研究过程中所确定的性能,应采取修正措施.
这些基本的质量控制满足在日常测试不确定度评估中使用确认方法研究的数据的主要要求.
在因果图中插入额外影响因子"重复性",计算opP的模型变为:psampleecrefoprefopopFmRIVcIFPRe6hom10=1mgkg公式A4.
1这就是说,象对待均匀性一样,重复性用一个乘法因子pFRe来表示.
选择这样的形式是为了计算方便,就象以下所述.
现在考虑不同的影响因素.
1.
U精密度研究对不同面包样品中的典型有机磷农药进行一系列平行测试(同一均匀样品、完整的萃取/测定程序),以获得该分析程序的总的随机变化(精密度).
测定结果见表A4.
2.
标准化的差值数据(差值除以平均值)提供了总的随机变化的量度.
为了得到单次测量的相对标准不确定度的估计值,求该标准化差值的标准偏差并除以2,这样就可以把成对差值的标准偏差校F(hom)图A4.
6使用方法确认研究的数据后重新安排的因果图重复性m(样品)V(op)稀释V(ref)m(ref)l(ref)l(op)校准t(op)V(ref)m(ref)稀释校准温度校准校准线性c(ref)纯度(ref)V(op)V(op)校准湿度回收率l(ref)校准m(样品)m(gross)校准线性校准线性P(op)CNAS-GL06:2006第81页共148页2006年06月01日发布2006年07月01日实施正为单次测定值的标准不确定度.
这就给出了总的测试程序的随机变化的标准不确定度,包括回收率的随机变化,但不包括样品非均匀性影响,此标准不确定度为:27.
02/382.
0=.
注:初一看,平行测试提供的自由度可能不够.
但是做以上测试并不是为了得到特定种类的面包表A4.
2:农药分析平行测试的结果农药残留D1(mgkgP-1P)D2(mgkgP-1P)平均值(mgkgP-1P)D1与D2的差值差值/平均值马拉硫磷1.
301.
301.
300.
000.
000马拉硫磷1.
300.
901.
100.
400.
364马拉硫磷0.
570.
530.
550.
040.
073马拉硫磷0.
160.
260.
21-0.
10-0.
476马拉硫磷0.
650.
580.
620.
070.
114PirimiphosMethyl0.
040.
040.
040.
000.
000甲基毒死蜱0.
080.
090.
085-0.
01-0.
118PirimiphosMethyl0.
020.
020.
020.
000.
000甲基毒死蜱0.
010.
020.
015-0.
01-0.
667PirimiphosMethyl0.
020.
010.
0150.
010.
667甲基毒死蜱0.
030.
020.
0250.
010.
400甲基毒死蜱0.
040.
060.
05-0.
02-0.
400PirimiphosMethyl0.
070.
080.
75-0.
01-0.
133甲基毒死蜱0.
010.
010.
100.
000.
000PirimiphosMethyl0.
060.
030.
0450.
030.
667中一种特定农药的分析测试程序的精密度的准确数据.
以上测试更重要的是测试一系列不同的物质和不同的样品中所选择的有代表性的典型有机磷农药的含量.
可通过对许多材料的平行测试这种最有效的方法来达到,该方法(重复性估计)中每一种材料平行测试的自由度约为1.
2.
U偏差研究分析程序的偏差是通过在实验室内部确认研究中使用加料样品(将均匀的样品分份,其中的1份加料)来进行研究的.
表A4.
3收集了各种类型加料样品样品的长期研究的结果.
相关的线框填入有关的"面包"类型,显示了42个样品的平均回收率为90%,标准偏差为(s)为28%.
标准不确定度采用平均值的标准偏差:0432.
042/28.
0)eR(==cu显著性检测是用来确定平均回收率是否与1.
0有显著性差异.
检测统计数据t用下式来计算:CNAS-GL06:2006第82页共148页2006年06月01日发布2006年07月01日实施315.
20432.
0)9.
01()eR(eR1===cuct这个数据与95%置信度,n-1自由度的双边临界值critt比较(其中n是用来评估ceR的测试结果的数目),假如t大于或等于critt值,则ceR与1有显著性差异.
021.
231.
241,≥=crittt在这个例子中,使用了校正因子(ceR1),因此ceR被明确地包括在结果的计算中.
表A4.
3:农药残留的回收率研究物质残留类型浓度(mgkgP-1P)NP1)P平均值P2)P(%)SP2)P(%)废油PCB10.
08849黄油OC0.
653310912动物饲料1OC0.
325100909动物和蔬菜油脂1OC0.
3334102241987芸苔OC0.
323210418面包OP0.
13429028甜面包干OP0.
13308427肉和骨饲料OC0.
32589512玉米麸质OC0.
3259929油菜饲料1OC0.
325118913小麦饲料1OC0.
32525889黄豆饲料1OC0.
325138519大麦饲料1OC0.
32598422(1)测试次数.
(2)平均值和样品标准偏差s以回收率百分比给出.
3.
U其它不确定度来源图A4.
7的因果图表明哪一个其它不确定度来源是:(1)充分地地包括在精密度数据中;(2)包括在回收率中;(3)必须进一步研究,并最终在不确定度计算中加以考虑.
所有的天平和重要的体积测量装置处于常规的控制下.
精密度和回收率研究已经考虑了体积测量装置校准的影响,因为在研究中使用不同的容量瓶和移液管.
持续超过了半年时间的大量的随机变化研究,也包括了环境温度对结果的影响.
因此只剩下参考物质的纯度、GC响应中可能的非线性(在图CNAS-GL06:2006第83页共148页2006年06月01日发布2006年07月01日实施中用校准项refI和opI表示)和样品的均匀性这些不确定度分量需要研究.
制造商提供的标准物质的纯度为99.
53%±0.
06%.
纯度是潜在的不确定度来源,其标准不确定度为00035.
03/0006.
0=(矩形分布).
但是这个分量的贡献如此之小(如与精密度评估相比),所以很明显可以忽略这个分量.
在给定的浓度范围内有关的有机磷农药的响应线性已在方法确认研究中加以确认.
此外,在表A4.
2和表A4.
3中所给类型的不同含量测试研究中,非线性对观察到的精密度有贡献,因此不需要额外考虑.
实验室内部确认方法研究已经证明了这一点.
面包子样品的均匀性是最后剩下的不确定度来源.
尽管已经进行了大规模的文献检索,但还是没有有关面包产品中痕量有机成分分布的文献数据.
(初一看,这很令人惊讶,但大多数分析家总是尽量使样品均匀,而不愿意单独对非均匀性做出评估),而且直接测定均匀性也是不实际的.
因此可以根据使用的取样方法来评估这个不确定分量.
为了有助于评估,考虑了一系列可能的的农药残留分布.
并使用简单的二项式统计分布来计算被分析样品中农药总量的标准不确定度(见A4.
6节).
农药残留分布和最终样品中农药含量的相对标准不确定度计算结果如下:·农药残留仅分布在样品最表面:0.
58·农药残留均匀地分布在样品表面:0.
20·农药残留均匀地分布在整个样品中,但是因为蒸发和分解的原因,越靠近表面农药残留的浓度越低:0.
05-0.
10(取决于"表面层"的厚度).
分布(a)特别适于比例取样或者是完全均匀的:装饰性添加物(全谷物)加到一个表面上会是这种分布.
分布(b)被认为可能是最差的情况,分布(c)被认为最有可能,但是不易与(b)区别.
基于此,选择0.
20这个数值.
注:非均匀性模型的更详细的细节见本例的最后一节.
A4.
5步骤4:;计算合成标准不确定度在分析程序的实验室内部确认研究中,重复性、偏差和所有其它可能的不确定度来源已被彻底研究.
它们的数值和不确定度收集在表A4.
4中.
因为在数学模型(等式A4.
1)中都是相乘关系,因此这些数值可以被合成为:opopcopopcPPuPPu*==++=34.
0)(34.
02.
0048.
027.
0)(222CNAS-GL06:2006第84页共148页2006年06月01日发布2006年07月01日实施本例的电子表格形式(表A4.
5)见4.
5.
注意电子表格计算的是标称修正结果1.
1111的绝对值的不确定度(0.
373),给出的值是相对标准不确定度0.
373/1.
111=0.
34三个不同的不确定度分量的相对大小可用直方图来比较,图A4.
8所示的值),(ixyu取自表A4.
5.
重复性是测量不确定度的最大分量.
由于这个分量是从方法总的变异性得出的,可以进一步实验查明哪些可以改进.
例如,在取样前均匀一整条面包可以显著地降低不确定度.
扩展不确定度U(PBopB)是合成不确定度乘以包含因子2得到:opopoppppU*=**=68.
0234.
0)((1)重复性在分析程序的变异性研究中加以考虑(公式A4.
1中的FBRepB).
(2)在分析程序的偏差研究中加以考虑.
(3)在其它来源的不确定度评价中加以考虑.
A4.
6特殊部分:建立有机磷农药不确定度的非均匀性模型假设样品中所有有关的物质,无论其状态如何,均可以萃取出来进行分析,非均匀性最糟糕的情况是所有待测的物质全部集中在样品的一个或几个部分.
一个常见却紧密联系例子是在整个样品中的不同的部分有两个含量的待测物质,如LB1B和LB2B.
如果是随机抽样,这类非均匀性的影响可使用二项式统计法评估.
所需的数值是:在切开样品后随机选取的n个等份样品中待测物质含量的平均值μ和标准偏差σ.
表A4.
4:农药分析的不确定度F(hom)(3)图A4.
7不确定度其它来源的评估重复性(1)m(样品)V(op)稀释V(ref)m(ref)l(ref)l(op)校准(3)l(op)V(ref)m(ref)稀释校准(2)温度(2)校准(2)线性c(ref)纯度(ref)V(op)V(op)回收率(2)l(ref)校准(3)m(样品)m(gross)线性线性校准(2)校准(2)温度(2)校准(2)校准(2)CNAS-GL06:2006第85页共148页2006年06月01日发布2006年07月01日实施描述数值x标准不确定度u(x)相对标准不确定度u(x)/x评述重复性(1)1.
00.
270.
27不同类型样品的平行测试偏差(回收率)(2)0.
90.
0430.
048加料样品其它来源(3)(均匀性)1.
00.
20.
2基于假设模型的评估U(PBopB)/PBopB0.
34相对标准不确定度表A4.
5:农药分析的不确定度ABCDE1重复性偏差均匀性2数值1.
00.
91.
03不确定度0.
270.
0430.
245重复性1.
01.
271.
01.
06偏差0.
90.
90.
9430.
97均匀性1.
01.
01.
01.
289PBopB1.
11111.
41111.
06041.
33310u(y,xBiB)0.
30-0.
05070.
22211u(y)P2P,u(y,xBiB)P2P0.
14200.
090.
002570.
049381213u(PBopB)0.
377(0.
377/1.
111=0.
34,相对标准不确定度)各个参数的值输入到第二行C2到E2.
它们的标准不确定度在其下行(C3:E3).
电子表格把C2-E2的数据复制到均匀性偏差重复性P(op)00.
10.
20.
30.
4u(y,xB1B)(mgГP1P)u(y,xB1B)=(y/xB1B)·u(xB1B)的数值取自表A4.
5图A4.
8农药分析的不确定度CNAS-GL06:2006第86页共148页2006年06月01日发布2006年07月01日实施第二列B5到B7.
使用这些值计算出的值在B9(=B5*B7/B6,基于公式A4.
1).
C5是C2加上C3其不确定度后的重复性的值.
使用C5:C7的数据计算得到的结果在C9.
在D和E列遵循相同的程序.
第10行(C10:E10)的数据是第9行(C9:E9)的数据减去B9得到的.
第11行数据(C11:E11)是第10行(C10:E10)的数据平方,把(C11:E11)相加得到B11.
B11平方根得到B13,这是合成不确定度.
这些数值如下:22112211)()(nlllnplplpn+=+=μμ[1]221112)()1(llpnp=σ[2]其中1l和2l是从整个样品中总量为X含总体分数分别为1L和2L区域中抽取样品中待测物质的含量;1p和2p是从那些区域中选择含有等份样品的概率(与可选择的总体等份样品数量相比,n必须要小).
以上的数值可用以下的方式计算,假设一个典型的面包样品,大约12*12*24cm,每一小等份样品的尺寸为2*2*2cm(总共有432个等份样品),假设从其中随机选择15个等份样品并均匀.
例(a)待测物质限于样品的大的单个表面(顶表面).
因此2L是0,2l也是0;1L是1,每一份含有顶表面的等份样品就含有物质含量1l,根据给出的尺寸,显然等份样品的6分之1(2/12)满足该标准,因此1p是1/6或0.
167,1l是x/72(也就是有72份"顶表面"等份样品),可以给出:58.
044.
108.
208.
2)17.
01(167.
0155.
2167.
0151212121211=====***==**=μσσσμRSDllllll注:为了计算在整个样品中的含量X,乘以432/15,得到x的平均值的估计值:xxlx=*=**=72725.
2154321这个结果是典型的随机取样;平均值的期望值正好等于总体的平均值.
对于随机取样,除了已在这里表示为σ或RSD的重复性变化外,对总的不确定度没有其他贡献.
例(b)物质均匀地分布在所有平面上.
遵循以上相似的讨论,并假设所有的表面等份样品包含相同的物质含量21,ll也是0,1p根据所给的尺寸计算如下:CNAS-GL06:2006第87页共148页2006年06月01日发布2006年07月01日实施63.
0)241212()2088()241212(1=******=p也就是1p是"外部"2cm样品的分数.
使用相同的假设,则2721xl=注:与例(a)相比数值有变化.
2.
087.
15.
35.
3)63.
01(63.
0155.
963.
01512221211===*==***==**=μσσσμRSDllllll例(c)由于蒸发或其它损失,物质含量在接近表面时减少至0.
这个例子可以简单地考虑为例(b)的反面,这样160,37.
011xlp==,这样:33.
087.
15.
35.
3)37.
01(37.
0156.
537.
0151212121211===*==***==**=μσσσμRSDllllll可是,假如损失延伸的深度小于所移取的等份样品的尺寸如所估计的,每小块都含有1l和2l,1l和2l都不为零.
例如,所有外部等份样品均含有样品的50%的"中心"部分和50%的"外部"部分.
2962121xlll=*=2222122222215.
3)()37.
01(37.
0156.
201537.
01515)(37.
015lllllllll=***==*+**=*+**=σμ给出RSD是:1.
87/20.
6=0.
09在当前模型中,待测物质损失的厚度为1cm的,考查典型的面包样品其面包皮厚度小于或等于1cm,把这作为待测物质损失的深度,(面包皮的形成抑制了该深度下待测物质的损失),据此例(c)的实际变化,将给出所得的值σ/μ不超过0.
09.
注:在本例中,因为非均匀性比抽取作均匀化的等份样品要小,不确定度的降低就多.
一般来说,这将导致对不确定度的减少.
据此,当较大量的内含物(例如含在面包块中颗粒)中含有不成比例的CNAS-GL06:2006第88页共148页2006年06月01日发布2006年07月01日实施待测物质的量时,就无需另外建立模型.
假如含在抽取作均匀化的等份样品中的此类内含物的概率足够大,则其对不确定度的贡献将不会超过上述各例已计算的值.
CNAS-GL06:2006第89页共148页2006年06月01日发布2006年07月01日实施例子A5:原子吸收光谱法测定陶瓷中镉溶出量概要目的用原子吸收光谱法测定陶瓷器皿中镉溶出量,使用的测量程序是经验方法BS6748.
测量程序图A5.
1的流程图列出了测定陶瓷器皿中镉溶出量的各个步骤.
被测量dmmg2temptimeacidVL0fffdaVcr=表A5.
1描述了各变量.
不确定度来源的识别有关的不确定度来源如图A5.
2的因果图所示.
不确定度来源的量化不同分量的大小列于表A5.
1中,分量之间的比较见图A5.
2中的直方图.
图A5.
1可浸取金属步骤表A5.
1:测定镉溶出量的不确定度描述值x标准不确定度u(x)相对标准不确定度u(x)/xCB0B浸取液中镉含量0.
26mglP-1P0.
018mglP-1P0.
069d稀释系数(如使用的话)1.
0P注1P0P注1P0P注1PVBLB浸取液体积0.
332l0.
0018l0.
0054aBVB容器的表面积2.
37dmP2P0.
06dmP2P0.
025fBacidB酸浓度的影响1.
00.
00080.
0008fBtimeB浸泡时间的影响1.
00.
0010.
001fBtempB温度的影响1.
00.
060.
06r每单位面积镉溶出量0.
036mgdmP-2P0.
0033mgdmP-2P0.
09注1:本例中没有用到稀释.
因此d正好为1.
0.
准备表面处理用4%V/V醋酸填充样品浸泡均匀搅拌浸泡液AAS测定AAS校准结果制备校准标准CNAS-GL06:2006第90页共148页2006年06月01日发布2006年07月01日实施图A5.
2测定浸出镉的不确定度来源图A5.
3测定浸出镉的不确定度校准曲线f(酸)f(时间)f(温度)填充温度校准读数结果r1长度2长度面积c(0)V(L)a(V)df(temp)f(time)f(tacid)a(V)V(L)c(O)r12034u(y,xB1B)=(y/xB1B).
u(xB1B)的数值取自表A5.
4u(y,xB1B)(mgdmP2P)*1000CNAS-GL06:2006第91页共148页2006年06月01日发布2006年07月01日实施例A5:原子吸收光谱法测定陶瓷中溶出的隔详细讨论A5.
1介绍这是评价一个经验方法的不确定度的范例,该经验方法(BS6748)为测定从陶瓷、玻璃、玻璃-陶瓷和搪瓷器皿中溶出的金属.
该方法使用原子吸收光谱仪(AAS)通过用4%(v/v)醋酸水溶液浸泡测定从陶瓷表面溶出的铅或镉的量.
从这个分析方法获得的结果仅与用相同方法得到的其它结果相比较.
A5.
2步骤1:技术规定英国标准BS6748:1986"陶瓷、玻璃、玻璃-陶瓷和搪瓷器皿中溶出的金属限量"给出了完整的测定程序,并形成被测量的技术规定.
下面给出的仅仅是大概描述.
A5.
2.
1仪器和试剂的技术规定影响不确定度的试剂规格:·新配制的4%(v/v)冰醋酸水溶液,用水将40ml冰醋酸稀释至1升.
·4%(v/v)醋酸溶液中铅标准溶液的浓度为1)11000(±mgl.
图A5.
4浸取金属程序·4%(v/v)醋酸溶液中镉标准溶液的浓度为1)5.
0500(±mgl.
实验用玻璃仪器要求至少是B级,并且在测定过程中在4%醋酸溶液中不会溶出可检测到含量的铅或镉.
原子吸收光谱仪要求其检测限为:铅0.
2mglP-1P,镉0.
02mglP-1P.
A5.
2.
2程序图A5.
4图示说明了整个测定程序.
影响不确定度评估的技术规定:ⅰ)样品在(22±2)℃的条件下放置,适当时("类别1"的产品),测量样品的表面积.
如在本例中,表面积是2.
37dm(表A5.
1和A5.
3列出了本例的实验数据).
ⅱ)将(22±2)℃的4%v/v醋酸溶液倒入经预处理的样品中,使溶液填充的高度为距离样品溢出处1mm,可从样品上端边缘处测量,或者距离样品的平端或斜边的最边缘处6mm.
ⅲ)记录使用4%v/v醋酸溶液的量,精确至±2%(本例使用了332ml醋酸).
准备表面处理用4%V/V醋酸填充样品浸泡均匀搅拌浸泡液AAS测定AAS校准结果制备校准标准溶液CNAS-GL06:2006第92页共148页2006年06月01日发布2006年07月01日实施ⅳ)样品在(22±2)℃的条件下放置24小时(测镉时要放置在黑暗中),并采取适当的措施防止挥发损失.
ⅴ)放置后,搅拌溶液使其足够均匀,取一部分测试样,必要时进行稀释(稀释系数为d),选用适当的波长在AA仪器上进行分析,本例中是最小二乘法校准曲线.
ⅵ)计算结果,报告在总浸取液中铅和/或镉的量,对于类别1的产品,用每平方分米表面积含多少毫克铅或镉的方式表示,对于类别2和3的产品,用每升体积含多少毫克铅或镉的方式表示.
注:通过邮寄可从BSI客户服务部获取BS6748:1986完整的复印件,地址:389ChiswickHighRoad,LondonW44ALEngland.
电话:+44(0)2089969001.
A5.
3步骤2:识别和分析不确定度来源步骤1描述了"经验方法".
如果这个方法在指定范围内使用,方法的偏差被定义为零.
因此偏差的评估与实验室的操作有关,而与方法固有的偏差无关.
因为还没有一个有证标准物质用于这个标准方法,偏差的总体控制与影响结果的方法参数的控制有关.
这些影响量是时间、温度、质量和体积等.
稀释后醋酸溶液中铅或镉的浓度CB0B用原子吸收光谱法测定,计算公式如下:1000)(BBAC=1lmg其中:CB0B:在浸取液中铅或镉的浓度[mglP-1P]AB0B:浸取液中金属的吸光度BB0B:校准曲线的截距BB1B:校准曲线的斜率对于本例所考虑的类别1的产品,经验方法要求结果用每单位面积溶出的铅或镉的质量r来表示,r的计算式如下:dBaBAdaVCrVVL==100L0)(V2dmmg其附加参数是:r:每单位面积溶出的铅或镉的质量r[2dmmg]VBLB:浸取液的体积[l]CNAS-GL06:2006第93页共148页2006年06月01日发布2006年07月01日实施aBvB:容器的表面积[]2dmd:样品的稀释系数上述被测量公式的前面部分被用于绘制基本因果图(图A5.
5).
图A5.
5初步因果关系图对于本经验方法,目前尚无有证标准物质用于评估实验室的操作.
因此所有可能的影响量都要考虑,如温度、浸泡时间和酸度.
为了考虑附加的影响量,公式中需加入各自的修正因子,因此扩大为:temptimeacidVLfffdaVcr=0这些修正因子包含在已修正的因果图中(图A5.
6).
图中显示为影响cB0B的因素.
注:该标准所允许的温度范围会引入不确定度,这是由于被测量技术规定不完善而产生.
符合经验方法以及实际可行时,考虑温度的影响允许对被报告的结果范围进行评估.
尤其要注意由不同操作温度引起的结果变化,因为是在按照技术规定要求测试而得到的结果,因此不能合理地认为是偏差.
5.
4步骤3:量化不确定度来源这个步骤的目的是对先前识别的每一个不确定度的来源进行量化.
可以用实验数据或基于良好的假定来进行量化.
U稀释系数d校准曲线填充温度校准读数结果r1长度2长度面积c(0)a(V)dV(L)CNAS-GL06:2006第94页共148页2006年06月01日发布2006年07月01日实施对于本例,无需稀释浸取液,因此不用考虑其对不确定的影响.
U体积UVBLB图A5.
6添加了隐含假设(修正因子)的因果关系图填充体积:经验方法要求容器被溶液填充至"距离边缘1mm以内".
对于典型的饮用和厨房用具,1mm将代表器皿高度的1%.
因此容器被填充的体积为99.
5±0.
5%(即大约是容器体积的0.
995±0.
005).
温度:醋酸的温度必须在22±2℃,由于与容器相比液体具有更大的体积膨胀,这个温度范围导致体积测量的不确定度.
假定温度分布为矩形分布,则332ml体积的标准不确定度是:ml08.
032332101.
24=***读数:记录体积VBLB要求的准确度在2%范围内,实际上使用量筒时允许约1%的不准确性(即0.
01VBLB).
假定是三角形分布来计算标准不确定度.
校准:体积校准是根据制造商的技术规格进行的,500ml量筒有±2.
5ml的偏差,假设为三角形分布,计算标准不确定度.
本例中体积为332ml,四个不确定度分量按下式合成:mlVuL83.
1)65.
2()633201.
0(08.
0)6332005.
0()(2222=+*++*=校准曲线f(酸)f(时间)f(温度)填充温度校准读数结果r1长度2长度面积c(0)a(V)dV(L)CNAS-GL06:2006第95页共148页2006年06月01日发布2006年07月01日实施U镉浓度UcB0使用手工绘制的校准曲线计算溶出镉的量.
用1)5.
0500(±mgl镉标准溶液中配制五个标准溶液,其浓度分别为0.
1、0.
3、0.
5、0.
7和0.
9mglP-1P.
使用线性最小二乘法拟合曲线程序的前提是假定横坐标的量的不确定度远小于纵坐标的量的不确定度,因此通常的cB0B不确定度计算程序仅仅与吸光度不确定度有关,而与校准溶液不确定度无关,也不与从同一溶液中逐次稀释产生必然的相关性.
然而在本例中,校准标准溶液的不确定度足够小以至可以忽略.
五个校准标准溶液分别被测量三次,结果见表A5.
2.
表A5.
2:校准结果浓度(mglP-1P)1230.
10.
0280.
0290.
0290.
30.
0840.
0830.
0810.
50.
1350.
1310.
1330.
70.
1800.
1810.
1830.
90.
2150.
2300.
216表A5.
3:镉溶出量分析的中间值和不确定度描述数值标准不确定度u(x)相对标准不确定度u(x)/xcBoB浸取液中镉的含量0.
26mglP-1P0.
018mglP-1P0.
069VBLB浸取液的体积0.
332l0.
0018l0.
0054aBVB器皿的表面积2.
37dmP2P0.
06dmP2P0.
025fBacidB酸浓度的影响1.
00.
00080.
0008fBtimeB浸泡时间的影响1.
00.
0010.
001fBtempB温度的影响1.
00.
060.
06校准曲线为:ABj:B第BiB个校准标准溶液的第j次吸光度BBCAiij0+=CNAS-GL06:2006第96页共148页2006年06月01日发布2006年07月01日实施Ci:第i个校准标准溶液的浓度BB1B:斜率BB0B:截距值标准偏差BB1B0.
24100.
0050BB0B0.
00870.
0029图A5.
7两次测量的线性最小二乘法拟合和不确定度区间线性最小二乘法拟合曲线的相关系数r为0.
997.
拟合曲线见图A5.
7.
残余标准偏差S是0.
005486.
测量浸出溶液两次,浓度cB0B为0.
26mglP-1P,附录E3详细说明了与线性最小二乘法拟合曲线程序有关的不确定度u(cB0B)计算.
因此这里只是简述不相同的计算步骤.
xxScCnPBScu200)(11)(++=2.
1)5.
026.
0(15121241.
0005486.
02++=10018.
0)(=mglCu残差标准偏差S为:0.
250.
200.
150.
050.
00.
00.
20.
40.
60.
81.
0吸光率鋀的浓度[mg/l]CNAS-GL06:2006第97页共148页2006年06月01日发布2006年07月01日实施005486.
02)]([2101=+=∑=nCBBASjnjj以及2.
1)(1==∑=njjxxcCS其中:BB1B:斜率P:测试cB0B的次数n:测试校准溶液的次数CB0B:浸出液中镉的浓度c:不同校准标准溶液的平均值(n次)i:下标,指第几个校准溶液j:下标,指获得校准曲线的测量次数U面积aUBUVUB长度测量:测量样品容器的尺寸计算其总的表面积为2.
37dmP2P,因为样品近似于圆筒但不规则,在95%置信水平中测量偏差估计在2mm范围内.
典型的尺寸介于1.
0dm和2.
0dm之间,其估计的尺寸测量不确定度为1mm(95%的数值除以1.
96后).
典型的面积测量需要高和宽两个长度尺寸(即1.
45dm和1.
64dm).
面积:由于样品没有完整的几何形状,因此面积计算也有不确定度,在本例中,在95%置信水平时估计有另外5%的分量.
长度测量和面积测量的不确定度分量按通常方式合成.
22296.
137.
205.
001.
001.
0)(*++=vau206.
0)(dmauv=U温度影响UfBtempB已进行了温度对陶瓷器皿溶出金属影响的一些研究P(1-5)P.
一般来说,温度影响是相当大的并且随着温度变化,溶出金属呈指数级上升趋势,直至达到极限值.
只有一个研究P1P给出20-25℃温度范围的影响.
从图形资料上看,在接近25℃的温度附近金属溶出量随温度的变化近于线性,其斜率约为5%℃P-1P.
经验方法允许±2℃的范围导致温度系数fBtempB为1±0.
1.
假定为矩形分布,将其转换为标准不确定CNAS-GL06:2006第98页共148页2006年06月01日发布2006年07月01日实施度:06.
031.
0)(==tempfuU时间影响fUBUtimeU对于相对较慢的过程,如浸泡过程,溶出量将大约与时间的微小变化成正比.
Krinitz和Franco发现在浸泡过程的最后6个小时中浓度的平均变化在86mglP-1P时大约是1.
8mglP-1P,即约占0.
3%/小时,因此对于(24±0.
5)小时的浸泡时间.
CB0B需要用系数UfUBUtimeUB进行修正:1±(0.
5X0.
003)=1±0.
0015.
这是矩形分布,产生的标准不确定度为:001.
030015.
0)(=timefuU酸浓度UfBacidB有一个研究酸浓度对铅溶出影响的结果显示,当浓度从4%改变为5%v/v时,某一特定批陶瓷的铅溶出量从92.
9变为19.
101mgl,acidf变为097.
09.
929.
929.
101=或近似0.
1.
而另一个研究使用热浸泡方法,结果显示类似的变化(浓度从2%变为6%v/v时,铅含量有50%的改变).
假定这个影响近似于与酸浓度成线性,估计酸浓度的每变化1%v/v,fBacidB约有0.
1的变化.
在另一个实验中,使用标准NaOH滴定的滴定法建立了该酸浓度和它的标准不确定度()vvuvv/008.
0/%996.
3=,.
采用该酸浓度的不确定度为0.
008%v/v,则fBacidB的不确定度为0008.
01.
0008.
0=*,因为该酸浓度的不确定度已表示为标准不确定度,这个值可被直接作为与fBacidB有关的不确定度.
注:原则上,不确定度值需要对上述单个研究是足够代表所有陶瓷情况的假定进行修正,当然,目前的数据已给出了对不确定度数量的合理评估.
A5.
5步骤4:计算合成标准不确定度假定没有稀释,则单位面积镉溶出量为:temptimeacidvLfffdaVcr=02dmmg中间值和标准不确定度被收集于表A5.
3,将这些数据代入:2036.
00.
10.
10.
137.
2332.
026.
0=****=mgdmr为了计算相乘表示式(见上)的合成标准不确定度,将标准不确定度的每个分量代入下式:CNAS-GL06:2006第99页共148页2006年06月01日发布2006年07月01日实施22222200)()()()()()()(+++++=temptemptimetimefacidacidvvLLcffuffuffuaauVVuCCurru095.
006.
0001.
00008.
0025.
00054.
0069.
0222222=+++++=20034.
0095.
0)(==mgdmrruc表A5.
4给出了计算合成标准不确定度的更简单的电子表格方法,附录E给出了该方法的详细说明.
图A5.
8图示了不同参数和影响量对测量不确定度的贡献,将每个分量的大小(表5.
4中的C13至H13)与合成不确定度(B16)进行比较.
扩展不确定度UB(r)B通过使用包含因子2计算得到:UBrB=0.
0034X2=0.
007mgdmP-2因此按照BS6748:1986标准测量镉溶出量为:(0.
036±0.
07)mgdmP-2上述不确定度计算使用的包含因子为2.
图A5.
8溶出镉测定的不确定度A5.
6范例5的参考资料1,B.
Krinitz,V.
Franco,J.
AOAC56869-875(1973)2,B.
Krinitz,J.
AOAC61,1124-1129(1978)3,J.
H.
Gould,S.
W.
Butler,K.
W.
Boyer,E.
A.
Stelle,J.
AOAC66,610-619(1983)4,T.
D.
Seht,S.
Sircar,M.
Z.
Hasan,Bull.
Environ.
Cintam.
Toxicol>10,51-56(1973)5,J.
H.
Gould,S.
W.
Butler,E.
A.
Steele,J.
AOAC66,1112-1116(1983)+f(temp)f(time)f(tacid)a(V)V(L)c(O)r12034u(y,xB1B)=(y/xB1B).
u(xB1B)的数值取自表A5.
4u(y,xB1B)(mgdmP2P)*1000CNAS-GL06:2006第100页共148页2006年06月01日发布2006年07月01日实施表A5.
4:镉溶出量分析的不确定度电子表格法计算ABCDEFGH10cLVVaacidftimeftempf2数值0.
260.
3322.
371.
01.
01.
03不确定度0.
0180.
00180.
060.
00080.
0010.
06450c0.
260.
2780.
260.
260.
260.
260.
266LV0.
3320.
3320.
33380.
3320.
3320.
3320.
3327Va2.
372.
372.
372.
432.
372.
372.
378acidf1.
01.
01.
01.
01.
00081.
01.
09timef1.
01.
01.
01.
01.
01.
0011.
010tempf1.
01.
01.
01.
01.
01.
01.
061112r0.
0364220.
0389430.
0366190.
0355230.
0364510.
0364580.
03860713),(ixyu0.
0025210.
000197-0.
0008990.
0000290.
0000360.
0021851422),()(ixyuyu,1.
199E-56.
36E-63.
90E-88.
09E-78.
49E-101.
33E-94.
78E-61516)(ruc0.
0034在第二行从C2到H2输入参数值,它们的标准不确定度在下一行(C3:H3),电子表格将C2:H2值复制到第二列B5:B10处,使用这些数值后的结果(r)值在B12中,C5显示了CB0B的值,等于C2加上其在C3的不确定度,使用C5:C10值计算的结果在C12中给出,D列和H列按照类似的步骤,13行(C13:H13)显示了C12:H12行减去B12值的结果,在14行(C14:H14)是13行(C13:H13)的值的平方,其和的结果在B14中给出,B16是合成标准不确定度,它是B14的平方根.
CNAS-GL06:2006第101页共148页2006年06月01日发布2006年07月01日实施例A6:动物饲料中粗纤维的测定概要目的用法定的标准方法测定粗纤维.
测试程序测试程序是一个标准的程序,图A6.
1列出了一般步骤.
对空白样进行重复测试以获得空白修正.
被测量纤维含量以与样品重量的百分比表示:acbCfibre100)(*=a:样品质量(约1g)b:测试过程中灰化后的质量损失(g)c:空白测试中灰化后的质量损失(g)不确定度来源的识别图A6.
9给出了完整的因果图.
图A6.
1纤维测试不确定度分量的量化实验室的实验表明在实验室内部,实验方法以可以采用协同研究的复现性数据的方式执行.
通常没有其它显著分量.
在低含量时需要考虑所使用的特定干燥程序.
对结果不确定度的典型评估列入表A6.
1内(以标准不确定度表示).
表A6.
1:合成标准不确定度纤维含量(%w/w)标准不确定度u(CBfibreB)(%w/w)相对标准不确定度u(CBfibreB)/CBfibreB2.
531.
0115.
029.
022=+0.
1250.
40.
08100.
60.
06研碎和称量样品加酸消化加碱消化干燥并称量残留灰化并称量残留结果CNAS-GL06:2006第102页共148页2006年06月01日发布2006年07月01日实施例A6:动物饲料中粗纤维的测试详细讨论A6.
1介绍在方法范围中粗纤维被定义为不溶于酸和碱介质的不含脂肪的有机物.
程序是标准化的,其结果可直接使用,改变程序就改变被测量,因此这是经验方法的一个例子.
该法定方法已有协同研究数据(重复性和复现性).
所述的精密度实验已计划作为方法性能内部评估的一部分.
本方法没有合适的标准物(即被相同方法所验证).
A6.
2步骤1:技术规定对于许多分析方法,被测量的技术规定最好是即全面描述分析方法的不同阶段,并提供被测量的公式.
程序从消化、过滤、干燥、灰化和称重的复杂程序,在图A6.
2中均有简要说明.
对空白坩埚的重复试验也使用该程序.
目的是消化大多数的组分,而留下所有不被消化的物质.
有机物被灰化,留下无机残留物,干燥的有机/无机残留物重量与灰化后残留物重量之差就是"纤维含量".
主要步骤如下:ⅰ)研磨样品使其通过1mm筛子;ⅱ)称量1g重样品置于已称重的坩埚中;ⅲ)加入规定浓度和体积的酸消化试剂,煮沸一定时间(按规定),过滤和冲洗残渣;ⅳ)加入标准碱消化试剂,煮沸一定时间,过滤,用丙酮冲洗;ⅴ)在标准规定的温度下干燥至恒重("恒重"在公布的方法中没有定义;也没有给出其它的干燥方法,如空气循环或残留物的分散);ⅵ)记录干燥残渣的重量;ⅶ)在规定温度下灰化至"恒重"(实际上在内部实验研究后决定灰化的时间);ⅷ)称量灰化残渣的重量,减去空白坩堝中残留物重量后计算纤维的含量.
被测量纤维含量以占样品重量的百分比表示:acbCfibre100)(*=a:样品重量(g),称取约1g样品来分析CNAS-GL06:2006第103页共148页2006年06月01日发布2006年07月01日实施b:测试过程中灰化后的重量损失(g)c:空白测试中灰化后的重量损失(g)图A6.
2动物饲料中纤维测定法规方法步骤流程图A6.
3步骤2:识别和分析不确定度来源识别不确定度各种来源,以该方法因果图进行说明(见图A6.
9),这个图按照附录D的程序被简化并去掉重复的部分.
加上去掉不显著的分量,得到A6.
10简化的因果图.
因为本方法有以前协同研究和内部研究的数据,使用这些数据与评估不确定度分量有密切关系,所以下面做进一步的讨论.
A6.
4步骤3:不确定度分量的量化协同试验结果研磨样品使其通过1mm筛子称量坩埚作空白验称1g样品置于坩埚中加助滤剂、防泡沫剂,接着加入150ml沸腾HB2BSOB4B剧烈沸腾30分钟过滤并用3*30ml沸水冲洗加入防泡沫剂,接着加入150ml沸腾KOH剧烈沸腾30分钟过滤并用3*30ml沸水冲洗使用真空装置,用3*25ml丙酮冲洗在130°C干燥至衡重在475~500°C灰化至衡重计算粗纤维含量%CNAS-GL06:2006第104页共148页2006年06月01日发布2006年07月01日实施该方法是协同试验项目,在协同试验研究中分析了五种不同的代表典型的纤维和脂肪含量的饲料.
协同研究的参加者按本方法的全部过程进行了试验,包括样品的研磨.
从协同试验中获得的重复性和复现性估计值见表A6.
2.
作为方法内部评估的一部分,实验计划用与协同试验所分析样品纤维浓度类似的饲料进行重复性(批精密度)评估,结果见表A6.
2,每一个内部重复性评估是基于5次重复实验.
表A6.
2:方法的协合研究和内部重复性检查的结果汇总纤维含量(%w/w)协同试验结果内部重复性标准偏差样品平均值复现性标准偏差重复性标准偏差A2.
30.
2930.
1980.
193B12.
10.
5630.
3580.
312C5.
40.
3900.
2640.
259D3.
40.
3470.
2320.
213E10.
10.
5750.
3910.
327内部得到的重复性估计值与协同试验获得的相当,表明在这个特定实验室中的方法精密度与参加协同试验的实验室的方法精密度相近,因此在评估方法的不确定度中可以使用协同试验的复现性标准偏差.
为了评估不确定度,我们要考虑是否有未被协同试验包括的其它影响因素需要指出来.
协同试验包括不同样品基体和样品前处理,因为参加者得到的样品需要在分析测试前研碎.
与基体影响和样品前处理有关的不确定度不需要另外的考虑,其它影响结果的参数与方法中使用的萃取和干燥条件有关,这些要分别研究以保证实验室偏差得到控制(与复现性标准偏差比较很小).
要考虑的参数在下面讨论.
灰化的质量损失由于本方法没有合适的标准参考物质,通过考虑与方法每一个步骤有关的不确定度来进行内部偏差的评估.
有几个因素对灰化后重量损失有关不确定度有贡献:酸浓度碱浓度CNAS-GL06:2006第105页共148页2006年06月01日发布2006年07月01日实施酸消化时间碱消化时间干燥温度和时间灰化温度和时间试剂浓度和消化时间在以前发表的文章中已研究了酸浓度、碱浓度、酸消化时间和碱消化时间的影响,在这些研究中,评估了该参数变化对分析结果的影响,对于每个参数计算了灵敏系数(最终结果变化与参数变化的比率)和参数的不确定度.
表A6.
3中不确定度比表A6.
2中复现性数值小,例如对于含有2.
3%w/w纤维的样品,其复现性标准偏差为0.
293%w/w,与酸消化时间变化有关的不确定度评估值为0.
021%w/w(2.
3*0.
009),因此我们可以安全地忽略掉与这些方法参数的变化有关的不确定度.
表A6.
3与方法参数有关的不确定度参数灵敏度相关系数P注1P参数的不确定度最终结果的不确定度RSDP注4P酸浓度0.
23(mollP-1P)P-1P10013.
0mollP注2P0.
00030碱浓度0.
21(mollP-1P)P-1P10023.
0mollP注2P0.
00048酸消化时间0.
0031minP-1P2.
89minsP注3P0.
0090碱消化时间0.
0025minP-1P2.
89minsP注3P0.
0072注1:通过制作纤维含量变化与试剂浓度或消化时间的关系图评估灵敏度相关系数.
使用线性回归来计算分析结果随参数变化的变化率.
注2:从制备酸碱溶液所使用的容量瓶的精密度和准确性的估计值以及温度影响等来计算酸和碱溶液浓度的标准不确定度,例子A1-A3中有计算该溶液浓度不确定度的进一步例子.
注3:方法规定消化时间为30分钟,消化时间控制在±5分钟范围内,这是一个矩形分布,除以3后换算为标准不确定度.
注4:以相对标准偏差表示最终结果的不确定度,通过将参数的不确定度乘以其灵敏度相关系数计算得到.
干燥温度和时间没有以往的数据可用.
方法要求样品在130℃温度下干燥至"恒重".
本例中样品在130℃温度下干燥3小时然后称重.
再重复干燥一个小时并称重.
在这个实验室中恒重被定义为连续两次称重的CNAS-GL06:2006第106页共148页2006年06月01日发布2006年07月01日实施变化小于2mg.
在内部实验研究中,四种饲料的平行样在110、130、150℃温度下干燥3和4小时后称重.
在大多数情况下,3小时和4小时之间的样品重量变化小于2mg,因此被作为干燥引起重量变化的不确定度的最坏情况进行评估,±2mg的范围为矩形分布.
除以3被转化为标准不确定度,因此干燥至恒重后所记录重量的不确定度是0.
00115g,方法规定样品重量为1g,对于1g样品,干燥至恒重的不确定度对应于0.
115%w/w纤维含量的标准不确定度.
这个不确定度来源独立于样品中纤维含量,因此不管样品中纤维浓度是多少,对于每一个样品的不确定度评估都将有固定的0.
115%w/w的分量.
对于所有纤维含量,这个不确定度小于复现性标准偏差,除了很低纤维浓度以外,都小于SBRB值的1/3,所以这个不确定度来源通常可以忽略.
对于低纤维浓度,这个不确定度大于SBRB值的1/3,因此在不确定度评估中要给予额外考虑(见表A6.
4).
表A6.
4:合成标准不确定度纤维含量(%w/w)标准不确定度u(CBfibreB)(%w/w)相对标准不确定度uu(CBfibreB)/CBfibreB2.
531.
0115.
029.
022=+0.
1250.
40.
08100.
60.
06灰化温度和时间方法要求样品在475℃至500℃温度下灰化至少30分钟,已经公布的关于灰化条件影响的研究,该研究涉及在450℃30分钟到650℃3小时之间的一系列不同灰化温度/时间条件下纤维含量的测量.
在不同条件下获得的纤维含量没有显著性差异,因此灰化温度和时间的小的变化对最终结果的影响可被忽略掉.
空白灰化后质量损失对于这个参数没有实验数据可用,然而正如上面所讨论的,这个参数变化产生的影响很小.
A6.
5步骤4:计算合成标准不确定度这是有协同试验数据的经验方法的实例.
内部重复性已进行了评估,并且与协同试验所预测的相当,因此可以使用从协同试验得到的sBRB值.
步骤3的讨论可以得出这样的结论,除了低纤维浓度下干燥条件的影响之外,所识别的其它不确定度来源与sBRB值比都很小,在这类情况下,可根据从协同试验获得的复现性标准偏差sBRB值进行不确定度评估.
对于纤维含量为2.
5%w/w的样品,应附加考虑与干燥条件有关的不确定度.
CNAS-GL06:2006第107页共148页2006年06月01日发布2006年07月01日实施标准不确定度表A6.
4给出了一定范围的纤维浓度典型的标准不确定度.
扩展不确定度表A6.
5给出了典型的扩展不确定度,计算时使用包含因子为2,置信水平约为95%.
表A6.
5:扩展不确定度纤维含量(%w/w)扩展不确定度U(CBfibreB)(%w/w)扩展不确定度CV(%)2.
50.
622550.
816101.
212CNAS-GL06:2006第108页共148页2006年06月01日发布2006年07月01日实施粗纤维(%)干燥温度干燥时间空白灰化后重量损失(c)样品重量(a)精密度灰化前坩埚重量天平线性天平校准坩埚重量碱消化P*P酸消化P*P灰化温度灰化时间天平校准天平线性坩埚重量天平校准天平线性样品称量精密度萃取精密度灰化精密度称量精密度灰化后样品和坩埚的重量灰化温度灰化时间天平线性天平校准消化条件萃取时间消解密酸消化酸体积酸浓度干燥温度干燥时间天平线性天平校准消化条件萃取时间沸腾速率碱消化酸体积酸浓度灰化前样品和坩锅的重量灰化后重量损失(b)*加到"酸消化"和碱消化"上的分支为清楚起见省略掉了,因为影响它的因素与影响样品的因素相同(即消化条件、酸浓度等).
图A6.
9动物饲料中纤维测定的因果关系图灰化后坩埚重量坩埚重量坩埚重量CNAS-GL06:2006第109页共148页2006年06月01日发布2006年07月01日实施样品称量精密度萃取灰化精密度称量精密度粗纤维(%)干燥温度干燥时间空白灰化后重量损失(c)精密度灰化前坩埚重量碱消化P*P酸消化P*P灰化温度灰化时间灰化后样品和坩埚的重量灰化温度灰化时间消化条件萃取时间沸腾速率酸消化酸体积酸浓度干燥温度干燥时间消化条件萃取时间凝胶速率碱消化酸体积酸浓度灰化前样品和坩锅的重量灰化后重量损失(b)*加到"酸消化"和碱消化"上的分支为清楚起见被省略掉了,因为影响它的因素与影响样品的因素相同(即消化条件、酸浓度等).
图A6.
10简化的因果关系图灰化后坩埚重量CNAS-GL06:2006第110页共148页2006年06月01日发布2006年07月01日实施例子A7使用双同位素稀释和电感耦合等离子体质谱测定水中的铅含量A7.
1介绍本例说明了在使用同位素稀释质谱(IDMS)和电感耦合等离子体质谱(ICP-MS)技术时,如何应用不确定度测定水样中铅的含量.
双IDMS概述:IDMS是被Comitèconsultatifpourlaquantetèdematière(CCQM)认可的一种可能成为基准的测量方法.
因此它有如何计算被测量的详细表达式.
作为最简单的例子,用具有富集同位素参考物质的标定示踪物进行同位素稀释,测量示踪样、样品和已知样品量与示踪样的混合物b中的同位素率.
下式给出了样品中元素的含量xc:∑∑=iyyixxxxbbbbyyxyyxiiiiRKRKRKRKRKRKmmcc)()(1111(1)其中:xc和yc分别是样品和示踪样的元素含量(用符号c代替k代表含量是为了避免与K-因子和包含k因子同符号而引起的混乱).
xm和ym是样品和示踪样的质量,yxRR,和bR是同位素含量比率.
下标x,y和b分别代表样品、示踪样和混合物.
选择一种通常在样品中最丰富的一种同位素,所有其它同位素的含量表示为它的相对比率.
选择一对特定的同位素(参考同位素和在示踪样中含量最丰富的同位素),作为监测比率,如)(/)(206208pbnpbn.
ixR和iyR分别是样品和示踪样中所有可能的同位素含量比率.
对于参考同位素,这个比率是1.
iiyxKK,和bK是对样品、示踪样和混合物中具体同位素含量比率的质量甄别校正因子.
使用标定的同位素标准物质,根据公式(2)测定K因子.
biasKKK+=0;其中observedcertifiedRRK=0(2)其中0K是时间为0时的质量甄别校正因子,在测量时,当K因子用于校正不同时间测量的校正比率时,biasK作为偏差因子就开始生效.
biasK还包括其它的可能的偏差,如乘数死时间校正,基体效应等.
certifiedR是同位素标准物质证书上的标定的同位素含量比率,observedR是该同位素标准物质观测值.
CNAS-GL06:2006第111页共148页2006年06月01日发布2006年07月01日实施在IDMS实验中,使用电感耦合等离子体质谱(ICP-MS),质量碎片随时间改变,因此要求对公式(1)中所有的同位素含量比率分别作质量甄别校正.
富含一种特定同位素的标定标准物质常常是难以获得的.
为了克服这个问题,经常使用"双"IDMS.
程序是使用一个没有很好确定特性,但是富含同位素示踪样的物质与一个具有天然同位素组成的标定标准物质(标识为z)联合使用.
此标定的、天然组成的物质作为基准定量分析标准品.
使用两种混合物,混合物b是样品和富含同位素示踪样的混合物,如公式(1)所示.
为了进行"双"IDMS,第二个混合物b′是由含量为1zc的基准分析标准品与富集物质为y制备而成.
这里给出了与公式(1)相近的表达式:∑∑=iyyizzzzbbbbyyzyyziiiiRKRKRKRKRKRKmmcc).
().
(.
.
.
.
1111'''''(3)其中:zc是基准分析标准溶液的元素含量,zm是制备新的混合物时的基准分析标准的质量,'ym是富含同位素示踪样溶液的质量.
1,,''zbbKRK和1zR分别是新的混合物和基准分析标准物的K因子和同位素比率.
下标z表示该基准分析标准.
将公式(1)除以公式(3)得:∑∑∑∑=iyyizzzzbbbbyyzyyiyyixxxxbbbbyyxyyzxiiiiiiiiRKRKRKRKRKRKmmcRKRKRKRKRKRKmmccc)()()()(11111`111'''''(4)化简这个公式,引入分析程序中的空白cBblackB,得到:blankizzixxbbyyzzbbxxbbbbyyyzxyzxcRKRKRKRKRKRKRKRKRKRKmmmmcciiii*=∑∑)()('''''11111111(5)这是最终的一个公式,在其中yc已经被消掉.
在本测量中,同位素含量的比率R的下标数字代表了以下实际同位素含量比率:)(/)()(/)(20620732062081pbnpbnRpbnpbnR==)(/)()(/)(20620442062062pbnpbnRpbnpbnR==作为参考,所有参数汇总在表A7.
1中.
CNAS-GL06:2006第112页共148页2006年06月01日发布2006年07月01日实施A7.
2步骤1:技术规定测量的总的步骤见表A7.
2中.
所涉及的计算和测量在下面说明.
表A7.
1IDMS参数概述参数描述参数描述mBxB混合物b中样品的质量(g)mByB混合物中富集标准样品的质量(g)mP'PByB混合物b'中富含同位素标准样品的质量(g)mBzB混合物b'中基准分析标准的质量(g)cBxB样品x的含量(molgP-1P或molgP-1P)P注1PcBzB基准分析标准z的含量(molgP-1P或molgP-1P)P注1PcByB标准样品y的含量(molgP-1P或molgP-1P)P注1PcBblankB程序空白中所观察的含量(molgP-1P或molgP-1P)P注1PRBbB所测得的混合物b的比率,n(P208PPb)/n(P206PPb)KBbBRBbB的质量偏差校正因子RP'PBbB所测得的混合物b'的比率,n(P208PPb)/n(P206PPb)KP'PBbBR'BbB的质量偏差校正因子RBy1B所测得的富含同位素的标准样品中富含同位素与标准同位素的比率KBy1BRBy1B的质量偏差校正因子RBziB基准分析标准中的所有比率,RBz1,BRBz2B等KBziBRBziB的质量偏差校正因子RBxiB样品中所有的比率KBxiBRBxiB的质量偏差校正因子RBx1B所测得的样品x中富集同位素与标准同位素的比率RBz1B同RBxi,B但是在基准分析标准中注1:含量的单位通常在文件中说明.
表A7.
2总程序步骤描述1制备基准分析标准2制备混合物b'和b3测定同位素比率4计算样品中Pb的含量cBxB5评估cBxB的不确定度含量xc的计算程序在这个测定水中铅的过程中,对每个混合物b'(定量分析标准+标准样品)和b(样品+标准样品)CNAS-GL06:2006第113页共148页2006年06月01日发布2006年07月01日实施各制备了四份混合样.
这就给出了4个xc值.
每一次测量按表A7.
2步骤1至4所述的进行.
报告值cBXB是4次重复测试的平均值.
计算摩尔质量由于某些元素(如Pb)的同位素组成的天然变化,将会对含量zc产生影响,所以必须测定基准分析标准的摩尔质量M.
注意若zc以1lgmo为单位那就没有影响.
对于一个元素E,其摩尔质量M(E)在数值上等于元素E的原子量)(EAr,原子量可以根据通式进行计算:∑∑===piipiiirREMREA11)()((6)其中iR是元素E所有的真实的同位素含量比率.
)(EMi是列表的核素质量.
注意公式(6)中的同位素含量比率必须是绝对的比率,也就是,它必须用质量甄别进行修正.
使用合适的下标得到公式(7).
在进行计算时,核素质量)(EMi取自文献数值2,而比率izR和0K-因子)(ziKo要进行测定(见表A7.
8),得到:11121034.
207.
)(.
.
)1,(====∑∑gmolRKEMRKAssaypbMpizzpiizzziiii(7)测定K-因子和同位素含量比率为了修正质量甄别,使用了公式(2)中所规定的修正因子K.
可使用有已标定同位素组成的标准物质计算0K-因子.
此种情况中,用同位素已标定的标准物质NISTSRM981监测0K-因子可能的变化.
0K-因子在修正比率前后进行测定.
一个典型的样品次序是:1.
(空白),2.
(NISTSRM981),3.
(空白),4.
(混合物1),5.
(空白),6.
(NISTSRM981),7.
(空白),8.
(样品)等.
空白测定不仅仅是用来做空白修正,也可以用来监测空白计数.
只有当空白样品计数率稳定并回到正常水平才能做新的测试.
注意在测试前应把样品、混合物、示踪样品和定量分析标准品稀释至适合的含量水平.
比率测定的结果、所计算的0K-因子和biasK的结果汇总在表A7.
8中.
基准分析标准的制备和含量zc的计算CNAS-GL06:2006第114页共148页2006年06月01日发布2006年07月01日实施制备两个基准分析标准,其中铅是从化学纯度为W=99.
999%的不同金属铅块上获得的.
两块铅块都来自同一批的高纯度铅.
将两个铅块样品溶于3/3:110wHNOwml:水中后微微加热,然后进一步稀释.
两个混合物各由这两个定量分析标准来制备.
下面介绍一个分析标准的一些数值.
将0.
36544g(1m)铅用).
05.
0(13llmHNO水溶液溶解并稀释至总重为14.
1961=dg.
此溶液定为标准1(Assay1).
取标准1质量为0292.
12=mg,用).
05.
0(13llmHNO水溶液稀释到总重为931.
992=d克,这个溶液定为标准2(Assay2).
标准2的Pb含量zc根据公式(8)计算如下:1181122lg092605.
0lg102605.
9)1Assay,(1=*==momopbMdwmdmczμ(8)混合物的制备示踪样品的质量分数是已知的,约为20μgPb/g的溶液.
也已知该样品中Pb的质量分数在这个范围内.
表A7.
3列出了本例中使用的两个混合物的称量数值.
表A7.
3混合物bb'所使用的溶液加料样品样品加料样品样品参数mBy1BmBxBmBy2BmBzB质量(g)1.
13601.
04401.
06541.
1029程序空白Blankc的测量在本例中用外标法测定程序空白,一个更复杂的方法是将富含同位素的示踪样品加入到空白中,并用与样品同样的方法处理它.
在本例中,只使用高纯度的试剂,这样会产生混合物的极端比率并随之产生富含同位素示踪程序的低可靠性.
外标校准程序空白被测定4次,得到Blankc值为17lg105.
4*moμ,按A类不确定度评估,其标准不确定度为17lg100.
4*moμ.
计算未知含量xc将测定和计算的数据(表A7.
8)代入公式(5)中得到1.
053738.
0=gmolcxμ.
四次重复测定值见表A7.
4.
A7.
3步骤2和3:不确定度来源的识别和量化CNAS-GL06:2006第115页共148页2006年06月01日发布2006年07月01日实施不确定度计算的思路假如把公式(2)、(7)和(8)代入最终的IDMS公式(5),参数数目之多使得公式无法处理.
为了使公式简单化,将0K-因子和标准定量分析溶液的含量和它们相关的不确定度分别处理,然后才代入IDMS公式(5).
在本例中,这样将不影响xc最终的总不确定度.
出于实际的考虑,公式做这样的简化是可行的.
为了计算总的标准不确定度)(xcuc,使用其中一次测量的数值(如在A7.
2中所述).
将使用附录E所描述的电子表格方法计算xc的总不确定度.
表A7.
4)lg(1moCxμ重复试验1(本例子)0.
053738重复试验20.
053621重复试验30.
053610重复试验40.
053822平均值0.
05370实验标准偏差(S)0.
0001K-因子的不确定度:i)0K的不确定度K用公式(2)计算得到,作为一个例子使用1xK的数值得出KB0B:9992.
01699.
21681.
2)(10===observedcertifiedRRxK(9)为了计算0K的不确定度,我们首先查看标准证书,证书给出的比率是2.
1681,在95%置信度时其不确定度为0.
0008.
为了把基于95%置信度的不确定度转化为标准不确定度,我们把以上的不确定度除以2得到标准不确定度0004.
0)(=certifiedRu.
观察到的数量比率,)(/)(206208pbnpbnRobserved=的标准不确定度为0.
0025(rsd).
K-因子的总不确定度计算如下:002507.
0)0025.
0()1681.
20004.
0()())((22110=+=xKxKuc(10)CNAS-GL06:2006第116页共148页2006年06月01日发布2006年07月01日实施这明显看出标准证书的比率对不确定的贡献是可以忽略的.
因此,所测量的比率observedR的不确定度将用作0K的不确定度.
biasK的不确定度:引入偏差因子(biasK)是因为考虑到质量甄别因子的数值可能的偏离.
正如在上述因果图和公式(2)中所见到的,每一个K-因子都有偏差.
这些偏差的数值在本例中是不知道的,所以取0.
当然每一个偏差均有不确定度,当计算最终不确定度时必须考虑这些偏差不确定度.
原则上,象公式(11)中使用的偏差一样,通过引用公式(5)以及参数1yK和1yR可以表示这个原理:KKLK+=1).
()((110ybiasxRyKyKc.
……(11)所有偏差值),,(zixiyiKbias在(0±0.
0001)之间.
该估计值是基于铅IDMS测试的长期经验,所有的),,(zixiyiKbias参数没有详尽地包括在表7.
5,表7.
8或公式5中,但是它们在所有不确定度计算中都被使用.
表A7.
5数值标准不确定度类型P注1PKBbiasB(zi)00.
001BRBZ1B2.
14290.
0054AKB0B(z1)0.
99890.
0025AKB0B(z3)0.
99930.
0035AKB0B(z4)1.
00020.
0060ARBz2B10ARBz3B0.
91470.
0032ARBz4B0.
058700.
00035AMB1B207.
9766360.
000003BMB2B205.
9744490.
000003BMB3B206.
9758800.
000003BMB4B203.
9730280.
000003BCNAS-GL06:2006第117页共148页2006年06月01日发布2006年07月01日实施注1:A类(统计评估)或B类(其它)所称质量的不确定度在本例情况下,称量是由一个专业的质量计量实验室进行的.
称量使用的程序是一种运用所校正的砝码和比较仪的高低射标技术.
该技术对每一个样品质量测量至少重复6次.
应用了浮力校正.
在本例中没有运用化学计算法和杂质修正.
称量证书上的不确定度作为标准不确定度,见表A7.
8.
标准分析溶液含量zc的不确定度:i)Pb原子量的不确定度:首先计算标准溶液[标准1(Assay1)]的摩尔质量的总不确定度,表A7.
5中的数值已知或已被测得.
根据公式(7),摩尔质量的计算如下:4433221144433322111)1,(zzzzzzzzzzzzzzzRKRKRKRKMRKMRKMRMRKAssayPbM++++++=(12)为了计算标准溶液中Pb摩尔质量的总标准不确定度,使用附录E中的电子表格模型.
每个比率和0K测量八次:使用附录E电子表格模型计算出的不确定度为10010.
0gmol,得出摩尔质量12103.
207)1,(=gmolAssayPbM,.
ii)测定zc中总标准不确定度的计算为了计算标准溶液中Pb含量zc的不确定度,使用表A7.
2的数据和公式(8).
称量不确定度从其证书中得到,见表A7.
3.
公式(8)中使用的所有参数及其不确定度见表A7.
6.
使用公式(8)计算zc含量.
根据附录D.
5,zc的总标准不确定度由计算得到:000028.
0)(=zccu.
其标准不确定度为1lg000028.
0moμ(用rsd表示为0.
03%)时,得到1lg092606.
0=moczμ.
使用附录E的电子表格模型,计算重复测试1的)(xccu.
重复测试1的不确定度评估将作为该测量的不确定度.
由于公式(5)的参数多,没有用电子表格显示出来.
参数的数值和其不确定度以及xc的总不确定度见表A7.
8.
表A7.
6数值不确定度铅块质量,mB1B(g)0.
365440.
00005CNAS-GL06:2006第118页共148页2006年06月01日发布2006年07月01日实施第一次稀释液的总质量dB1B(g)196.
140.
03取第一次稀释液质量mB2B(g)1.
02920.
0002第二次稀释液的总质量dB2B(g)99.
9310.
01金属铅块的纯度w(质量分数)0.
999990.
000005标准物质中Pb的摩尔质量M(gmolP-1P)207.
21040.
0010A7.
4步骤4:计算总标准不确定度表A7.
7显示四次重复测定的平均值和实验标准偏差.
数值取自表A7.
4和表A7.
8.
在IDMS和很多非日常分析中,测试程序的完整统计控制需要耗费无数的资源和时间.
检查一些不确定度来源是否已被忽略的好的方法是将A类评估得出的不确定度与四次重复测试的实验标准偏差相比较.
假如实验标准偏差大于按A类评估的不确定度分量,表明测定过程没有完全被理解.
作为一种近似,使用表7.
8的数据,通过取总的实验不确定度的92.
2%,计算A类评估的实验不确定度的总和,为1lg00041.
0moμ.
这个数值明显高于1lg00010.
0moμ的实验标准偏差(见表A7.
7).
这表明实验的标准偏差已被按A类评估的不确定度分量所包括,因此无需进一步考虑来自混合物制备的按A类评估的不确定度分量.
然而可能有与混合物制备有关的偏差.
在本例中,混合物制备中可能的偏差与主要不确定度来源相比时,可判定混合物制备的偏差是不显著的.
水样中Pb的含量为:1lg)00036.
005370.
0(±=mocxμ该结果可提供使用包含2因子时的扩展不确定度.
表A7.
7重复测定1重复测定1-4的平均值cBxB=0.
05374cBxB=0.
05370molgP-1Pu(cBxB)=0.
00018s=0.
00010P注1PmolgP-1P注1:此为实验标准不确定度,不是平均值的标准偏差.
例子A7参考资料1.
T,Cvita,Metrologia,1996,33,35-392.
G,AudiandA.
H.
Wapstra,Nuclearphysics,A565(1993)CNAS-GL06:2006第119页共148页2006年06月01日发布2006年07月01日实施表A7.
8参数不确定评估数值实验不确定度(注1)对总不确定度的贡献uBcB(%)最终不确定度(注2)对总不确定度的贡献uBcB(%)ΣKBbiasBB00.
001P注3P7.
20.
001P注3P37.
6cBzBB0.
0926050.
0000280.
20.
0000280.
8KB0B(b)A0.
99870.
002514.
40.
000889.
5KB0B(b')A0.
99830.
002518.
30.
0008811.
9KB0B(x1)A0.
99920.
00254.
30.
000882.
8KB0B(x3)A1.
00040.
003510.
00120.
6KB0B(x4)A1.
0010.
00600.
00210KB0B(y1)A0.
99990.
002500.
000880KB0B(z1)A0.
99890.
00256.
60.
000884.
3KB0B(z3)A0.
99930.
003510.
00120.
6KB0B(z4)A1.
00020.
00600.
00210mBxBB1.
04400.
00020.
10.
00020.
3mBy1BB1.
13600.
00020.
10.
00020.
3mBy2BB1.
06540.
00020.
10.
00020.
3mBzBB1.
10290.
00020.
10.
00020.
3RBbBA0.
293600.
0007314.
20.
00026P注4P9.
5R'BbBA0.
50500.
001319.
30.
0004612.
7RBx1BA2.
14020.
00544.
40.
00192.
9RBx2BCons.
100RBx3BA0.
91420.
003210.
00110.
6RBx4BA0.
059010.
0003500.
000120RBy1BA0.
000640.
0000400.
0000140RBz1BA2.
14290.
00546.
70.
00194.
4RBz2BCons.
100RBz3BA0.
91470.
003210.
00110.
6RBz4BA0.
058700.
0003500.
000120cBblankBA4.
5*10P-7P4.
0*10P-7P02.
0*10P-7P0cBxB0.
053740.
000410.
00018ΣABcontrib.
.
B=92.
2ΣBBcontrib.
.
B=7.
8ΣABcontrib.
.
B=60.
4ΣBBcontrib.
.
B=39.
6注1:实验不确定度在计算时没有考虑每个参数的测量次数.
注2:在最终不确定度的计算中考虑了测定次数.
在本例中所有按A类评估的参数测定了8次.
其标准不确定度要除以8.
注3:该数值是一个KBbiasB的数值.
用参数ΣKBbiasB来代替所有的KBbiasB(zi,xi,yi)的列表,这些数值都是(0±0.
001).
注4:每个混合物其RBbB测定8次,总共测定32次.
当没有混合物之间的变异时,如本例,在此模型中所有混合物均进行四次重复测定时,就能得出了所有32个测定值.
这要花费很多时间,在本例中因为它不会显著地影响不确定度,这就没有做.
CNAS-GL06:2006第120页共148页2006年06月01日发布2006年07月01日实施附录B定义总则B.
1测量准确度测量结果与被测量的真值之间的一致程度[H.
4].
注1:"准确度"是一个定性概念.
注2:不要用"精密度"代替"准确度".
B.
2精密度在规定条件所获得的独立测量结果之间的一致程度[H.
5].
注1:精密度只取决于随机误差的分布,而与真值或规定值无关.
注2:精密度的度量通常用不精密度术语表示,并计成测量结果的标准偏差.
大的标准偏差反映了小的精密度.
注3:"独立测量结果"意味着所获得的测量结果不受以前任何同样或类似物体的测量结果所影响.
定量测量精密度关键取决于规定的条件.
重复性和重现性条件就是一组规定的极端条件.
B.
3真值与给定的特定量的定义一致的值[H.
4].
注1:这是一个通过完善的测量才能获得的值.
注2:真值按其本性是不确定的.
注3:"真值"要与不定冠词"a"而不是定冠词"the"一起使用,因为与给定的特定量的定义一致的值可能有很多.
B.
4约定真值对于给定目的具有适当不确定度,赋予特定量的值,有时该值是约定采用的[H.
4].
例:a)在给定地点,由参考标准复现而赋予该量的值可作为约定真值.
b)常数委员会(CODATA)1986年推荐的阿伏加德罗常数值注1:"约定真值"有时称为指定值、最佳估计值、约定值或参考值.
注2:常用某量的多次测量结果来确定约定真值.
B.
5影响量不是被测量但对测量结果有影响的量[H.
4].
例1:用来测量长度的千分尺的温度;mol1100221367.
623*CNAS-GL06:2006第121页共148页2006年06月01日发布2006年07月01日实施2:交流电位差幅值测量中的频率;3:测量人体血液样品血红蛋白浓度时的胆红素的浓度.
测量B.
6被测量作为测量对象的特定量[H.
4].
注:对被测量的详细描述,可要求包括对其他有关量(如时间、温度和压力)作出说明.
B.
7测量以确定量值为目的的一组操作[H.
4].
B.
8测量程序进行特定测量时所用的,根据给定的测量方法具体叙述的一组操作[H.
4].
注:测量程序(有时被称为测量方法)通常记录文件中,并且足够详细,以使操作者在进行测量时不再需要补充资料.
B.
9测量方法进行测量时所用的,按类别叙述的一组操作逻辑次序[H.
4].
注:测量方法可按不同方式分类,如替代法、微差法、零位法.
B.
10测量结果由测量所得到的赋予被测量的值.
注:当给出"测量结果"时,应说明它是示值、未修正测量结果或已修正测量结果,还应表明它是否为几个值的平均值.
注2:在测量结果的完整表述中应包括测量不确定度的信息.
不确定度B.
11(测量)不确定度表征合理地赋予被测量之值的分散性,与测量结果相联系的参数.
注1:此参数可以是诸如标准偏差(或其指定倍数),或说明了置信水平的区间半宽度.
注2:测量不确定度由多个分量组成.
其中一些分量可用测量列结果的统计分布估算,并用实验标准偏差表征.
另一些分量则可用基于经验或其他信息的假定概率分布估算,也可用标准偏差表征.
注3:测量结果应理解为被测量之值的最佳估计,而所有的不确定度分量均对分散性有贡献,包括那些由系统效应引起的分量,如与修正值和参考标准有关的分量.
B.
12溯源性CNAS-GL06:2006第122页共148页2006年06月01日发布2006年07月01日实施通过一条具有规定不确定度的不间断的比较链,使测量结果或测量标准的值能够与规定的参考标准,通常是与国家测量标准或国际测量标准联系起来的特性[H.
4].
B.
13标准不确定度)(ixu:以标准偏差表示的测量结果ix的不确度[H.
2].
B.
14合成标准不确定度)(yuc:当测量结果y是由若干个其他量的值求得时,按其他各量的方差或协方差算得的标准不确定度[H.
2].
B.
15扩展不确定度确定测量结果区间的量,合理赋予被测量之值分布的大部分可望含于此区间[H.
2].
注1:该部分可认为是该区间包含概率或区间的置信水平.
注2:为了将特定的置信水平与扩展不确定度确定的区间联系起来,需要对表征测量结果及其合成不确定度的概率分布作出清晰或不容置疑的假设.
表征该区间的置信水平只能达到该假设所允许的合理程度.
注3:扩展不确定度U按下述公式由合成标准不确定度cu和包含因子k计算得到:cukU*=B.
16包含因子为求得扩展不确定度,对合成标准不确定度所乘之数字因子[H.
2].
注:包含因子一般在2-3范围内.
B.
17(不确定度的)A类评定用对观测列进行统计分析的方法,来评定标准不确定度[H.
2].
B.
18(不确定度的)B类评定用不同于观测列进行统计分析的方法,来评定标准不确定度[H.
2].
误差B.
19(测量)误差测量结果减去被测量的真值[H.
4].
注:由于真值不能确定,实际上用的是约定真值.
B.
20随机误差测量的结果与在重复性条件下,对同一被测量进行无限多次测量所得结果的平均值之差[H.
4].
CNAS-GL06:2006第123页共148页2006年06月01日发布2006年07月01日实施注1:随机误差等于误差减去系统误差.
注2:因为测量只进行有限次数,故可能确定的只是随机误差的估计值.
B.
21系统误差在重复性条件下,对同一被测量进行无限多次测量所得结果的平均值与被测量的真值之差[H.
4].
注1:系统误差等于误差减去随机误差.
注2:如真值一样,系统误差及其原因是不能完全获知.
统计术语B.
22算术平均值x:一个样品n个结果的算术平均值.
nxxnii∑==,1B.
23样品标准偏差s:由一个样品的n个结果得出的总体标准偏差σ的估计值.
1)(12=∑=nxxsniiB.
24平均值的标准偏差从总体中抽取n个数值,其平均值的标准偏差由下式给出:nssx=术语"标准误差"和"平均值的标准误差"也曾用来表达同样量.
B.
25相对标准偏差(RSD)RSD:由一个样品的n个结果得出总体标准偏差的估计值除以该样品的平均值,也称变异系数.
通常也用百分比表示:xsRSD=CNAS-GL06:2006第124页共148页2006年06月01日发布2006年07月01日实施附录C:分析过程中的不确定度C.
1为了识别分析过程中的不确定度可能来源,将分析过程分解成一组通用的步骤是有帮助的:1:抽样2:样品制备3:有证标准物质对测试系统的影响4:仪器的校准5:分析(数据采集)6:数据处理7:结果的表达8:结果的解释C.
2这些步骤可按对不确定度的贡献进一步分组.
下面所列出的内容,虽然不一定全面,但提供应考虑因素的指南.
1.
抽样-均匀性-具体的抽样策略的影响(例如,随机抽样、分层随机抽样、比例抽样等)-媒介移动的影响(尤其是密度选择)-媒介的物理状态(固体、液体、气体)-温度和压力影响-抽样过程是否影响组成例如,在抽样系统中的差色吸附.
2.
样品制备-均匀性和/或二级抽样的影响-干燥-碾磨-溶解-萃取-污染-衍生(化学影响)-稀释误差CNAS-GL06:2006第125页共148页2006年06月01日发布2006年07月01日实施-(预)浓缩-物种形成影响的控制3.
有证标准物质对测量系统的影响-有证标准物质的不确定度-有证标准物质是否与样品匹配4.
仪器的校准-使用有证标准物质的仪器校准误差-标准物质及其不确定度-校准用的物质是否与样品匹配-仪器的精密度5.
分析-自动分析仪的进位-操作者的影响,例如色盲、视差、其他系统误差-基体、试剂或其他被分析物的干扰-试剂的纯度-仪器参数的设置,例如积分参数-重复性实验的精密度6.
数据处理-平均-修约的控制-统计-运算法则(模型拟合,例如线性最小二乘法)7.
结果的表达-最终结果-不确定度的估计-置信水平8.
结果解释-对照限值/范围-法规的符合性-目的的适用性CNAS-GL06:2006第126页共148页2006年06月01日发布2006年07月01日实施附录D:分析不确定度来源D.
1介绍通常有必要将分析方法有关的所有不确定来源分析出来并加以记录.
将这一过程系统化通常是有用的,既可保证考虑范围的全面性,又可避免重复过高.
下面的步骤(基于以前出版的方法[H.
14]),提供了一种合适的、系统地分析不确定度产生原因的可能方法.
D.
2方法的原理D.
2.
1该策略分成两步:·识别对结果的影响因素实际上,通过使用因果图(有时称作Ishikawa或"鱼骨"图)[H.
15]来进行必要的系统分析.
·简化并解决重复的情况首次列出的内容要进行精简并且保证影响因素没有不必要地重复列出.
D.
3因果分析D.
3.
1构造因果图的原理在其他地方加以详述.
所使用的步骤如下:1.
写出结果的完整公式.
该公式中的参数构成因果图的主要分支.
几乎有必要增加一个对总偏差(通常以回收率来表示)修正的主要分支.
适当时,推荐在此本步骤中增加此分支.
2.
考虑方法的每一步骤,并且从主要影响因素之外来考虑,在因果图上进一步增加其他因素,如环境及基体的影响.
3.
对每一个分支,增加有贡献的影响因素直至影响因素变得足够小,即直到对结果的影响可忽略.
4.
解决重复问题,并重新安置,澄清影响因素,将有关的有不确定度来源编成组.
在该步骤在单独的精密度分支上集合所有精密度内容是便利的.
D.
3.
2因果分析的最后步骤要求进一步说明.
对每个输入参数的贡献量进行详细分析时,自然会产生重复性问题.
例如,对任何影响因素,重复性实验的变异性总是存在的,至少在名义上.
这些影响因素总体上会对方法的总体方差有贡献.
因此,假如已有这样考虑了,就不需单独列出.
同样,通常用同一台仪器称量物质,会导致校准不确定度的重复计算.
出于这些考虑,就有了下述精简因果图的附加规则(虽然它完全等同地适用于任何系统地列出的影响因素).
·取消影响因素:两者均要去掉.
例如,在差减称量中,称量两次,两次均受天平"零偏差"的影响,"零偏差"将由于重量差而消除.
因此,可在分别列出的称量有关分支中取消.
·类似的影响因素,同样时间:合成一个单一输入量.
例如:许多输入量的重复性变化能合成一CNAS-GL06:2006第127页共148页2006年06月01日发布2006年07月01日实施个总的重复性精密度"分支".
尤其需要注意,每一次测量单独操作间的变异性可以合成,而对整批次操作间的变异性(例如仪器校准)只有用批次间精密度度量时才能观测到.
·不同的情况:重新标注.
通常会发现类似命名的影响因素实际上是指类似测量的不同情况.
在进行下一步之前,必须清楚区分.
D.
3.
3这种类型的分析不会导致单一结构的列表.
在目前的例子中,温度既可视为所测密度的直接影响因素,也可视为是对此比重瓶中的物质所测质量的影响因素,两者均可成为首次结构内容.
实际上这不影响方法的使用性.
假如所有重要的影响因素在列表的某个地方只出现过一次,总的一套方法仍然有效.
D.
3.
4一旦因果图分析完成,一种适当的做法是回到结果的原始公式,并增加任何新的项(例如温度)到公式中.
D.
4例子D.
4.
1本步骤通过参照简化了的直接密度测量例子来说明.
考虑直接测量乙醇密度d(EtOH)的例子,通过称量合适的带刻度容器的皮重mtare以及加了乙醇后的毛重mgross来获得已知体积乙醇的质量.
密度按下式计算d(EtOH)=(mgross-mtare)/V为了清晰,仅考虑三个影响因素:仪器校准、温度和每次测量的精密度.
图D1-D3用图表的方式说明了这过程.
D.
4.
2因果图由一个结果的分支结构组成,其最终只导致一个结果.
对目前的目的而言,该结果就是具体的分析结果(图D1的'd(EtOH)').
指向该结果的'各分支'是贡献因素,包括具体的中间测量结果和其他因素,诸如环境或基体影响.
每一个分支接着又有自己的贡献因素.
这些"因素"包含影响结果的各种因素,无论是变量或常数.
这些因素的不确定度都明显地对结果的不确定度有贡献.
D.
4.
3图D1显示了应用步骤1-3直接获得的一种可能的图表.
主要分支是公式中的参数,对各参数的影响因素由次分支来表示.
注意,有两个"温度"影响因素,3个精密度影响因素和3个"校准"影响因素.
D.
4.
4图D2显示了按照第二条规则(相同影响因素/时间)将精密度和温度各自组合在一起.
温度可作为影响密度的单一因素,而每次测量的变异性均贡献给整个方法的重复实验所观测到的变异性.
D.
4.
5按照第一个精简规则(取消),两个称量的校准偏差相互抵消了,可以去除(图D3).
D.
4.
6最后,余下的校准分支需要分成两个(不同)分量,一个可能是由于天平响应的非线性,另一个是与体积测量有关的校准不确定度.
线性=inearity精密度CNAS-GL06:2006第128页共148页2006年06月01日发布2006年07月01日实施图D1:首次列表温度d(EtOH)精密度m(毛)m(皮)线性偏差校准温度线性偏差校准校准体积精密度图D2:类似影响因素组合d(EtOH)温度m(毛)温度精密度线性偏差校准精密度体积校准精密度温度线性偏差校准m(皮)精密度图D3:取消d(EtOH)温度m(毛)线性偏差校准体积校准精密度线性偏差校准m(皮)偏差取消同一天平:CNAS-GL06:2006第129页共148页2006年06月01日发布2006年07月01日实施附录E:有用的统计程序E.
1分布函数下列表格显示了如何从两个最重要的分布函数的参数来计算标准不确定度,并给出它们能被使用的环境.
例:一个化学家估计一个影响因素不小于7或不大于10,并感到具体数值位于这个区间的任何地方,但不知道是否区间的任何部分比另一部分更加可能.
这是描述了区间2a=3(半宽a=1.
5)的矩形分布函数的情况.
使用下面矩形分布的函数,标准不确定度的估计值可计算出来.
使用上面的区间,a=1.
5,标准不确定度的结果87.
0)35.
1(=.
矩形分布图形在下述情况使用不确定度·证书或其他技术规定给出了界限,但无规定置信水平(例如:25ml±0.
05ml)·估计值是以最大区间(±a)形式给出的,但没给出分布的形状3)(axu=三角形分布图形在下述情况使用不确定度·所获得的有关x的信息不仅限于矩形分布.
靠近x的数值比接近两边界的更加可能.
·估计值是以最大区间(±a)形式作出并具有对称分布6)(axu=1/2ax2a(=±a)1/ax2a(=±a)CNAS-GL06:2006第130页共148页2006年06月01日发布2006年07月01日实施正态分布图形在下述情况使用不确定度估计值是对随机变化过程的重复测量作出的不确定度是以标准偏差s,相对标准偏差xs/或方差系数CV%给出,未给出分布不确定度以95%(或其他)置信水平,区间为x±c给出,未规定分布(95%置信水平)(99.
7%置信水平)E.
2电子表格方法计算不确定度E.
2.
1电子表格软件可用来简化第8节的计算.
该程序利用微分法的近似数字方法,并且只要求知道用来导出最终结果(包括任何必要的校正因子或影响)的计算以及参数的数值和它们的不确定度.
此处所描述的是按照Kragten[H.
12]的方法.
E.
2.
2在不确定度))xx,xu(y(n21K的表达式中假如y(xB1B,xB2B…xBnB)对ix是线性或与ix相比)(ixu是小的,偏导)/(xyi可近似为:乘以)(ixu获得因ix不确定度引起的y的不确定度),(ixyu,得:sxu=)(xCVxuxssxusxu===100%)()/()()(2/)(cxu=3/)(cxu=∑∑=+=nkikikixxuxyxynixiuxiy,1,2),(,1)()()())((xuxyxuxyxyiiiii+≈),,()))((,(),(2121xxxxyxxuxxxyxyuniniiiLLLL+≈1/ax2σCNAS-GL06:2006第131页共148页2006年06月01日发布2006年07月01日实施因此,),(ixyu只是分别用)]([iixux+和ix计算出来的y之差.
E.
2.
3线性或小的iixxu/)(的假设并不是在所有的情况下都充分得到满足.
尽管如此,当对评估)(ixu的值进行必要的近似进行考虑时,该方法确实提供对于实际用途的可接受的准确性.
参考文献H.
12对这点讨论更详细并且建议核查假设有效性的方法.
E.
2.
4基础的电子表格设立如下,假设结果y是四个参数p、q、r和s的函数.
ⅰ)在电子表格A栏内输入p、q等值以及计算y的公式.
将栏A中y的各变量复制到其他各栏(见图E2.
1).
如图所示将不确定度u(p)、u(q)等的值放在第一行是方便的.
ⅱ)将u(p)加到单元B3的p中,将u(q)加到单元C4的q中等等,见图E2.
2.
重新计算电子表格后,单元B8就变成),),((Lrqpupf+(在图E2.
2和E2.
3中用),,,'(Lrqpf表示),单元C8就变成),),(,(Lrquqpf+等.
ⅲ)在第九行输入第8行减A8(例如,单元B9变成B8-A8),给出u(y,p)的值为u(y,p)=f(p+u(p),q,r…)-f(p,q,r…)等ⅳ)为了得到y的标准不确定度,各个分量分别平方,并加在一起,然后开平方根,即通过在10行输入u(y,p)2(图E2.
3),并且将这些和的平方根放在A10.
即,单元A10相当于下列公式SQRT(SUM(B10+C10+D10+E10))它给出了y的标准不确定度.
E2.
5单元B10、C10等的内容显示了y不确定度的各个不确定度分量的平方分量u(y,xi)2=2))((iixuc,因此容易看出哪一个分量是显著的.
E2.
6随着个别参数值改变或不确定度的更新,可直接进行即时计算.
在上面的步骤i),不是直接将栏A复制到栏B至E中,而是通过引用而将p至s值复制,即单元3B至3E均引用3A、单元4B至4E引用4A等.
图2.
1的水平箭头表明第3行引用情况.
注意单元8B至8E还分别引用列B至列E的值,如图2.
1的B列竖箭头所示.
在上面的步骤ii)中,通过引用加上第一行的引用值(如图E2.
1箭头所示).
例如单元3B变成,13BA+单元4C变成14CA+等.
对参数或不确定度的改变将立刻在8A的总结果中及10A的合成标准不确定度中反映出来.
E2.
7假如变量是相关的,要在10A的SUM中增加必要的附加项.
例如,假如p和q相关,其相关系CNAS-GL06:2006第132页共148页2006年06月01日发布2006年07月01日实施数为),(qpr,则附加项),(),(),(2qyupyuqpr***要在开平方根之前加到所计算的和中.
因此通过电子表格中增加适当的附加项就可以很容易将相关性包括进去.
图E2.
1ABCDE1u(p)u(q)u(r)u(s)23ppppp4qqqqq5rrrrr6sssss78y=f(p,q…)y=f(p,q…)y=f(p,q…)y=f(p,q…)y=f(p,q…)91011图E2.
2ABCDE1u(p)u(q)u(r)u(s)23pp+u(p)ppp4qqq+u(q)qq5rrrr+u(r)r6sssss+u(s)78y=f(p,q…)y=f(p′…)y=f(…q′…)y=f(…r′…)y=f(…s′…)9u(y,p)u(y,q)u(y,r)u(y,r)1011CNAS-GL06:2006第133页共148页2006年06月01日发布2006年07月01日实施E.
3线性最小二乘法校准的不确定度E.
3.
1分析方法或仪器通常是通过观察被分析物x的不同浓度的响应值y来校准的.
在大多数情况下,这种关系被认为是线性的,即xbbyo1+=1.
3.
EEq利用该校准线,可通过样品中被分析物产生的响应值yBobsB,由下式测得其浓度xBpredB:1/)(bbyxoobspred=2.
3.
EEq通常通过对一组n对数值),(iiyx的加权或未加权最小二乘法回归来确定常数1b和obE.
3.
2为了获得估计值predx的不确定度,四种主要不确定度来源需要考虑.
·测量y时的随机变化,既影响标准响应值iy,又影响被测量的响应值obsy·导致标准值赋值ix误差的随机效应·ix和iy值可能受恒定的未知偏移的影响,例如当x值取自常备溶液的连续稀释时所产生的偏移·线性的假设未毕有效上面因素中,在正常操作中最显著的是y的随机变化,该种来源的不确定度评估方法将在此详述.
其它来源也简要地加以考虑以便指出所用的方法.
E.
3.
3由于y的(随机)变化性,预估值xpred的不确定度可按几种方法来评估:从计算所得的方差和协方差来获取.
假如1b和ob的值,它们的方差var(1b),var(ob)以及它们的协方差covar(obb,1)是由最小二乘法图E2.
3ABCDE1u(p)u(q)u(r)u(s)23pp+u(p)ppp4qqq+u(q)qq5rrrr+ur6sssss+u(s)78y=f(p,q…)y=f(p′…)y=f(…q′…)y=f(…r′…)y=f(…s′…)9u(y,p)u(y,q)u(y,r)u(y,s)10u(y)u(y,p)P2Pu(y,q)P2Pu(y,r)P2Pu(y,s)P2P11CNAS-GL06:2006第134页共148页2006年06月01日发布2006年07月01日实施获得,x的方差var(x)通过使用第八章提供的公式,并对该标准公式求微分,得:21112)var(),(cov.
.
2)var(.
)var()var(bbbbarxbxyxoopredpredobspred+++=Eq.
E3.
3以及对应的不确定度)var(),(predpredxyxu=从校准数据获得上述)var(predx的公式可用测量校准函数所使用的一组n个数据点),(iiyx来表示:++=∑∑∑∑)/)()(.
()(1.
/)var()var(22221221iiiiiprediobspredwxwxwxxwbSbyxEq.
E3.
4其中)(,)2()(22fiifiiiyynyywS=∑是第i个点的余差,n是校准的数据点的数目,1b是计算所得的最佳拟合斜率,iw是赋与iy的权以及)~(xxpred是predx与n个21,xx…值的平均值x之间的差.
对未加权数据以及)var(obsy是基于p次测量,公式E3.
4变成:++=∑∑)/)()(()(11.
)var(2212212nxxxxnpbSxipredpredEq.
E3.
5这是例子5所使用的公式:().
)(/)(222∑∑∑==xxnxxSiiixx由用来导出校准曲线的软件提供的信息某些软件给出S的值,不同叫法如RMS误差或残余标准误差.
然后可用在E3.
4或E3.
5中.
然而,一些软件也可从某些新的x值的拟合线上计算所得的y值的标准偏差)(cys,因此这可用来计算)var(predx,对于p=1()()∑∑++=nxxxxnysiipredc/)()(11)(222对照E3.
5,给出[]21/)()var(bysxcpred=Eq.
E3.
6E.
3.
4标准值ix各有自己的不确定度,并通过传播律传给最终结果.
实际上,这些数值的不确定度与系统的响应值iy的不确定度相比通常是小的,因此可以忽略.
由于具体的标准值的不确定度引起的预CNAS-GL06:2006第135页共148页2006年06月01日发布2006年07月01日实施估值predx的不确定度近似值为:nxuxxuiipred/)(),(≈EqE3.
7其中n是用于校准的ix值的数目.
这个表达式可用来检查),(ipredxxu的重要性.
E.
3.
5由于假设y和x的线性关系引起的不确定度通常不会大到要求做额外的评估.
假如余差没有表明与这种假设关系有重大系统偏差,由这种假设所引起的不确定度可忽略(除了导致y方差最终增加的假设外).
假如余差表明有系统偏离趋势,在校准函数有必要包括较高阶项式.
在这些情况下的var(x)的计算方法见标准文本.
基于系统偏离的大小也可做出判断.
E.
3.
6x和y的值可受恒定的未知偏移影响(例如当x的值取自给出不确定度的有证常备溶液的连续稀释时所引起的),假如这些影响对y和x产生的标准不确定度分别为u(y,const)和u(x,const),内插值predx的不确定度为)var()/),((),()(2122xbconstyuconstxuxupred++=Eq.
E3.
8E.
3.
7E.
3.
2所述的四个不确定度分量可用Eq.
E3.
3至Eq.
E3.
8的公式进行计算.
线性校准计算引起的总不确定度可按常规的方式合成这四个分量来计算.
E.
4与被分析物浓度有关的不确定度的表示E4.
1.
引言E4.
1.
1在化学测试中,经常观察到在被分析物浓度的大范围中,在总的不确定度中占支配作用的分量几乎与被分析物的浓度成比例变化,即)(xux∝.
在这类例子中,通常以相对标准偏差或如变化系数(%CV)来引述不确定度是合理的.
E4.
1.
2当不确定度不受浓度的影响,例如在低浓度或被分析物的浓度范围较窄时,引用不确定度的绝对值更合理.
E4.
1.
3在某些情况下,恒定的影响和按比例变化的影响都是重要.
当不确定度随被分析物浓度的变化是重要的以及简单作为一个变化系数报告是不合适时,本节给出了记录不确定度信息的通用方法.
E4.
2方法的基础E4.
2.
1既要考虑到不确定度的随被分析物浓度变化的比例性,又要考虑不随被分析物浓度变化的恒定数值的可能性,使用下列通用公式:CNAS-GL06:2006第136页共148页2006年06月01日发布2006年07月01日实施212).
()(sxsxuo+=[1]其中u(x)是结果x的合成标准不确定度(即,用标准偏差表示的不确定度)sB0B代表对总不确定度的贡献恒定的分量sB1B是比例常数该表达式是基于常用的将两个不确定度分量合成总不确定的方法,并假设分量(os)是常数,而分量)(1xs与结果成比例,图E4.
1展示了该表达式的形式.
注:上述方法只有当可能计算大量的数值时才可行.
当进行实验研究时,通常不能建立相关的抛物线关系.
在这种情况下,通过对在不同的被分析物浓度获取的四个或更多的合成不确定度进行简单的线性回归可获得合适的近似值.
该程序与根据ISO5725:1994进行的重现性和重复性的程序是一致.
有关的表达式为111.
)(sxsxuo+≈.
E4.
2.
2该图可分为大致的区域(见图A至C)A:不确定度受项0s的支配,因此基本恒定,约等于0sB:两项均相当重要.
最终的不确定度比0s或1xs高出许多,可见一部分曲线.
C项1xs占支配地位.
不确定度随x的增加而几乎呈线性增长,因此约等于1xs.
E4.
2.
3注意在许多实际例子中,没有明显的完整的曲线形式.
极常见的是,方法范围所允许的被分析物浓度的整个报告范围落在一个单一图区.
下面更详细介绍的一些特殊例子的结果就是这样.
E4.
3与浓度相关的不确定度数据的表示E4.
3.
1一般来说,不确定度可用0s和1s的每一个值的形式来表示.
在整个方法的范围内可用这些数值提供不确定度的估计值.
当在计算机系统上对描述完整的方法进行计算时,其中应用的通用公式独立于参数的值(其中之一可为0,见下面),这尤其重要.
因此建议,除下面给出的特定情况,或当相关性较强并且不是线性*之外,不确定度用以常量0s表示的数值和以变量1s表示的数值来表示.
*一个非线性相关的的重要例子是仪器噪音对接近仪器性能的上限高吸光度的吸光度测量的影响.
当吸光度是通过透射比率(如在红外光谱中)来计算时尤其明显.
在这种情况下,基线噪音使得高吸光度数值有非常大的不确定度,并且不确定度值增长地比简单的线性估计所预测地更快.
通常的做法是降低吸光度,尤其是通过稀释,使得吸光度的数值刚好落在工作范围内.
此处所使用的线性模型因此变得合适了.
其他例子包括一些免疫测定方法的"s形"响应.
CNAS-GL06:2006第137页共148页2006年06月01日发布2006年07月01日实施E4.
4特殊情况E4.
4.
1不确定度与被分析物的浓度无关(os起支配作用)不确定度通常会有效地独立于所观测的被分析物的浓度,当:·结果接近于零(例如在方法规定的检测限内).
图E4.
1的区域A.
·结果的可能范围(在方法的范围中规定的或在不确定度评估的范围声明中规定的)与所观测到的浓度相比是小的.
在这些情况下,1s的值可记为零.
0s通常是所计算的标准不确定度.
E4.
4.
2不确定度完全与被分析物的浓度相关(1s占支配作用)当结果远大于零(例如,高于"测量限")以及有明显的证据表明在方法范围所允许的被分析物浓度范围内,不确定度随着被分析物的浓度而按比例变化,1xs项起支配作用(见图E4.
1的区域C).
在这种情况下,以及方法范围不包括约为零的被分析物的浓度,0s可合理地记为零,1s是以相对标准偏差表示的不确定度.
E4.
4.
3中间相关在中间的情况,以及尤其当情况对应于图E4.
1区域B时,可采取两种方法:a)使用变量相关更常用的方法是测量、记录和使用0s和1s.
当需要时,不确定度的评估基于所报告的结果.
可行时推荐该方法.
注:见E4.
2注b)使用固定的近似在通常测试中可使用的另一个方法,当:·相关性不强(即,比例性的证据不强),或·所期望的结果范围是中等时.
上面任一情况均导致变化不超过平均不确定度估计值的15%的不确定度,因此基于所期望的结果平均值计算和引用一个不确定度的固定值作为通用值是合理的.
或者用x的平均值或特征值来计算固定的不确定度估计值,并且以此来代替个别计算的估计值,或基于研究覆盖所允许的被分析物浓度的全范围(在不确定度估计值的范围内)的物质研究,得到单一的标准偏差,并且几乎没有证据说明比例性的假设是合理的.
一般应该将它作为零相关的情况,有关的标准偏差记为s.
CNAS-GL06:2006第138页共148页2006年06月01日发布2006年07月01日实施E4.
5测定0s和1sE4.
5.
1在一个项式占支配作用的特殊情况下,分别以标准偏差或相对标准偏差表示的不确定度作为0s和1s的值通常是足够的.
然而,当相关性不甚明显时,有必要在不同的被分析物浓度间接地从一系列不确定度估计值来确定0s和1s.
E4.
5.
2假定从不同的分量进行合成不确定度的计算,其某些分量与浓度有关而另外一些则不相关,通常可通过模拟来研究总不确定度与被分析物浓度的相关性.
程序如下:1.
计算(或通过实验获取)涵盖所允许的全范围内至少十个被分析物浓度的不确定度;2.
画2)(ixu对2ix的图;3.
通过线性回归,获得cmxxu+=22)(的线中m和c的估计值;4.
从cs=0,ms=1计算0s和1s;5.
记录0s和1s.
E4.
6报告E4.
6.
1此处所列出的方法允许评估任何单次结果的标准不确定度.
原则上,当报告不确定度信息时,将以下述形式,[结果]±[不确定度]其中以标准偏差表示的不确定度按上述计算,并且假如必要时扩展(通常乘以因子2)以增加置信水平.
然而,当许多结果一起报告时,给出适合于所报告的所有结果的不确定度估计值是可能的并且完全可接受.
E4.
6.
2表E4.
1给出一些例子.
一系列不同被分析物的不确定度值可按同样的原则有效地计算.
注:当一个"检测限"或"报告限"用来给出以"
1几个例子的不确定度汇总场合占支配的项报告的例子所有结果的不确定度基本恒定0s或固定的近似值(E4.
4.
1或E4.
4.
3a节)标准偏差:扩展不确定度;95%置信水平的区间不确定度通常与浓度成比例1xs(见E4.
4.
2节)相对标准偏差:方差系数(%CV)比例性和不确定度低限值的混合中间例子(节E4.
4.
3)引用%cv或rsd以及标准偏差表示的低限sB0x.
sB1Bu(x)B1.
81.
61.
41.
210.
80.
60.
40.
20012345678不确定度约等于80不确定度比sB0B或x.
s.
都要大得多不确定度约等于x.
sABC结果x图E4.
1不确定度的观察结果的变化CNAS-GL06:2006第140页共148页2006年06月01日发布2006年07月01日实施附录F检测限/测量限的测量不确定度F.
1引言F.
1.
1在低浓度,日益增加的各种影响变得重要,包括,例如·噪音或不稳定基线的存在·干扰物对(毛)信号的干扰作用·所使用的任何分析空白的影响以及·在萃取、分离或净化过程中的损失因为这些影响,随着被分析物浓度降低,与结果有关的相对不确定度趋于增加,首先达到结果的很大一部分,最终达到其(对称的)不确定度区间包含零.
该区域是典型地与给定方法的实际检测限有关.
注:与测量和报告低浓度被分析物有关的术语和约定已经广泛在其他地方加以讨论(有关例子和定义见文献H.
16,H.
17,H.
18).
这里,术语"检测限"只表示检测成为疑问的一种浓度,并且不与任何特定的定义相关联.
F1.
2普遍认为,"检测限"最重要的用途是显示方法性能不足以提供可接受的定量,因此需要改进.
因此,理想时,定量测量不要在这个区域进行.
但是如此多的材料在这个低浓度是重要的,以至不可避免要在这个区域进行测量以及结果的报告.
F1.
3当结果是小的并且与结果相比不确定度是大的时,测量不确定度的ISO指南[H.
2]并没有给出不确定度评估的清晰指引.
确实,本导则第8章所描述的"不确定度传播律"的基本形式在这个区域可能不能精确使用.
该计算基于的假设之一是与被测量的值相比不确定度是小的.
从ISO指南给出的不确定度定义将产生另一个理论上的困难.
尽管在这个区域负的观察值是相当可能,并且甚至相当常见,当被测量是浓度时,所指的含零以下值的分散不能是".
.
.
.
.
.
合理赋予被测量的值",因为浓度本身不能为负.
F1.
4这些困难并未阻止本指南所描述的方法的使用.
但是在解释和报告该区域的测量不确定度结果时要谨慎.
本附录的目的是提供有限的指南补充从其他途径所获得的指南.
注:类似的考虑可适用于其他区域.
例如,摩尔或质量分数接近100%可导致类似的困难.
F.
2观测值和估计值F.
2.
1测量科学的基本原理是结果是真值的估计值.
例如,分析结果开始是以所观测到的信号为单位的,如mV,吸光度单位等.
为传递给更多的读者,尤其是实验室的客户或其他官方,原始数据需要转换成CNAS-GL06:2006第141页共148页2006年06月01日发布2006年07月01日实施化学量,如浓度或物质的量.
这种转换典型地要求校准程序(可包括,如对观测值的修正以及清楚描述的损失的修正).
然而,无论何种转换,所产生的数值仍为观测值或信号,假如实验是正确进行,该观测值数值仍为被测量的最佳估计值.
F2.
2观测值通常不被适用于实际浓度的同样基本限值所限制.
例如,报告一个所观测到的浓度,即低于零的估计值,是完全合理的.
谈到在同样区域内可能观察值的分散也是同样合理.
例如,当对一个没有被分析物存在的样品进行无偏见的测量时,人们应该看到约一半观察值低于零.
换句话说,类似的报告:观测到的浓度=1184.
2±mg观测到的浓度=1192.
4±mg不仅可能,它们应被视为有效的表达.
F2.
3本指南所描述的不确定度评估方法完全适合于观测值的不确定评估.
当报告观测值及其不确定度给一个内行的读者时,即使结果表明一个不可能的物理环境,对最佳值及其不确定度的报告的理解不会有阻碍和矛盾.
确实,在某些情况下(例如,当报告一个被用来修正其他结果的分析空白值时),报告该观测值及其不确定度(不管多大)是绝对必要的.
F2.
4这是真实的情况,无论结果的最终使用是否值得怀疑.
因为只有该观测值和它的不确定度能被直接使用(例如,在进一步计算、在趋势分析或重新解释中),未经审查的观测值应总是可获得.
F2.
5因此理想的情况是报告有效观测值及其不确定度,而不管其值如何.
F.
3经解释的结果和符合性声明F3.
1尽管前面所述的理由,为了最终用户的利益,必须承认许多包含一些解释的分析报告及符合性声明.
典型地,这类解释包括材料中合理存在的被分析物浓度有关的结论.
这类解释是关于真实情况的结论,因此被认为(被最终用户)是符合真正的限值.
因此,其任何不确定的估计值也是"真"值.
F3.
2当最终用途已被充分理解时,以及最终用户因实际上不能了解测量观测值的性质时,其它地方提供的有关报告低浓度结果的一般导则(例如文献H.
16,H.
17,H.
18)可合理地适用.
F3.
3然而进一步的谨慎是贴切的.
许多关于检测能力的文献依赖于重复性观测值的统计.
本指南的读者应该清楚所观察的变化只是罕有作为结果总不确定度的良好指南.
正如处理任何其他领域的结果一样,因此在报告的数值之前应仔细考虑影响该结果的所有不确定度.
CNAS-GL06:2006第142页共148页2006年06月01日发布2006年07月01日实施附录G不确定度的常见来源和数值下表概括了一些不确定度分量的典型例子.
该表给出·具体的被测量或实验程序(测定质量、体积等)·每一例中不确定度的主要分量和来源·测定每一个来源的不确定度所建议的方法·典型的例子该表旨在概括例子,并指出在分析中评估测量不确定度的一般方法.
它们并不旨在全面,所给的值也不可未经独立判断而直接使用.
然而,这些数值可帮助决定某个具体分量是否显著.
CNAS-GL06:2006第143页共148页2006年06月01日发布2006年07月01日实施典型值测量不确定度分量原因测量方法例子数值天平校准不确定度校准的有限准确度将校准证书上声明的值转换为标准偏差4位天平0.
5mg线性i)在有证砝码范围内实验ii)制造商的规格约0.
5*最后一位有效数字日偏移不同,包括温度长期核查称量的标准偏差.
必要时计算成RSD约0.
5*最后一位有效数字可读性显示器或刻度的有限分辨率来自最后一位有效数字0.
5*最后有效位/3重复性变化不同连续样品或核查称量的标准偏差约0.
5*最后一位有效数字密度影响(通常情况下)P注1P校准块/样品密度不配引起空气浮力效应的不同从已知或假设的密度和典型的空气条件来计算钠、镍、铝、有机固体、水、烃1ppm20ppm50-100ppm65ppm90ppm质量密度影响(在真空中)见上计算空气浮力影响并从校准块减去浮力影响100g水10g镍+0.
1g(效应)<1mg(效应)注1:对于基本常数或SI单位定义,通过称量测定质量通常修正到真空中的重量.
在大多数其他实际场合中,引用的重量是基于OIML所定义的常规情况[H.
18].
这个常规是引用在空气密度为32.
1kgm和样品密度为38000kgm的重量,相当于在正常大气条件下处于海平面称量钢.
当样品密度为38000kgm或空气密度为32.
1kgm时,对常规质量的浮力修正是零.
因为空气密度通常非常接近后者,对常规质量的修正通常可忽略.
上表所给出的在常规条件下称量的与密度有关的影响所产生的标准不确定度数值,对于在海平面常规情况下称量,且未考虑修正浮力的初步估计是足够的.
然而,在常规情况下测得的质量与"真实质量"(在真空中)相差0.
1%或更多(见上表最后一行的影响因素).
CNAS-GL06:2006第144页共148页2006年06月01日发布2006年07月01日实施典型值测量不确定度原因测量方法例子值校准不确定度校准有限的准确度制造商规格所声明的转换为标准偏差对ASTM体积为V的A类玻璃仪器,限值约为200/6.
0V10ml(A类)=ml01.
03/02.
0温度与校准温度不同引起的与在标准温度下的体积的差别给出相对标准偏差,其中ΔT是可能的温度范围,α是液体的体积膨胀系数,对于水,α约为14102*k;对于有机液体为13101*k100ml水在规定的操作温度3℃内进行测试为0.
03ml体积(液体)重复性变化不同连续核查排出的体积的标准偏差(通过称量决定)25ml移液管重复充满/称量s=0.
0092ml*假设矩形分布)32/(αΔTCNAS-GL06:2006第145页共148页2006年06月01日发布2006年07月01日实施典型值测量不确定度分量原因测量方法例子数值纯度不纯降低了标准物质的实际数量,活性不纯可干扰测量制造商证书所给出.
标准证书通常给出无法定量的限值.
因此可按矩形分布处理,除以3.
注:当不纯的性质未给出时,可需要额外的考虑或核查来建立干扰限标准邻苯二甲酸氢钾的证书值为99.
9±0.
1%%06.
03/1.
0=浓度(证书)证书给出的标准物质浓度不确定度制造商证书所给出.
标准溶液证书通常给出无法定量的限值;因此应按矩形分布处理,除以3在4%醋酸中的醋酸镉,证书值为1)21000(±mgl12.
132=mgl(RSD为)0012.
0标准物质浓度浓度(从有证材料配制)标准值和中间步骤的不确定度的合成合成前面步骤的数值作为整个过程的RSD三次稀释后醋酸镉:从11000mgl到15.
0mgl0034.
00017.
00021.
00017.
00012.
02222=+++作为RSD*假设为矩形分布CNAS-GL06:2006第146页共148页2006年06月01日发布2006年07月01日实施测量不确定度分量原因测量方法例子数值仪器校准注:该分量是指相对于标准吸光度的吸光度读数,而不是以吸光度的读数对浓度校准校准的有限准确度校准证书给出的作为限值,然后转换为标准偏差吸光度重复性化不同重复测量的标准偏差或QA性能7个吸光度读数的平均,s=1.
6362.
07/63.
1=抽样均匀性不均匀材料的二级抽样通常不能正确的代表整个物质.
注:随机抽样通常导致零偏差.
有必要检查抽样确实是随机的.
i)不同的二级抽样结果的标准偏差(假如与分析准确度比非均匀性较大)ii)从已知或假设的总体参数所估计的标准偏差从假设的2项不均匀性的面包中抽样(见例A4)从72份污染和360份未污染整体中抽样15份:RSD=0.
58CNAS-GL06:2006第147页共148页2006年06月01日发布2006年07月01日实施典型值测量不确定度分量原因测量方法例子值平均回收率萃取很少是完全的,并且可加入或包括干扰物回收率从相当的标准物质或代表性示踪的百分比回收率来计算.
从回收率实验的平均值的标准偏差可得不确定度注:回收率也可直接从以前测量所得的分配系数计算而得面包农药的回收率;42次实验,平均值为90%,s=28%(见例A4)%4342/28=(RSD为0.
048)萃取回收率回收率的重复性变化不同重复实验的标准偏差从成对平行试验的数据所得的面包农药的回收率(见例A4)RSD0.
31`CNAS-GL06:2006第148页共148页2006年06月01日发布2006年07月01日实施附录H参考文献H.
1.
ISO/IEC17025:1999.
GeneralRequirementsfortheCompetenceofCalibrationandTestingLaboratories.
ISO,Geneva(1999).
H.
2.
GuideToTheExpressionOfUncertaintyInMeasurement.
ISO,Geneva(1993).
H.
3.
EURACHEM,QuantifyingUncertaintyinAnalyticalMeasurement.
LaboratoryoftheGovernmentChemist,London(1995).
ISBN0-948926-08-2H.
4.
InternationalVocabularyofbasicandgeneraltermsinMetrology.
ISO,Geneva,(1993).
(ISBN92-67-10175-1)H.
5.
ISO3534:1993.
Statistics–VocabularyandSymbols.
ISO,Geneva,Switzerland(1993).
H.
6.
AnalyticalMethodsCommittee,Analyst(London).
12029-34(1995).
H.
7.
EURACHEM,TheFitnessforPurposeofAnalyticalMethods.
(1998)(ISBN0-948926-12-0)H.
8.
ISO/IECGuide33:1989.
UsersofCertifiedReferenceMaterials.
ISO,Geneva(1989).
H.
9.
InternationalUnionofPureandAppliedChemistry.
PureAppl.
Chem.
,67,331-343,(1995).
H.
10.
ISO5725:1994(Parts1-4and6).
Accuracy(truenessandprecision)ofmeasurementmethodsandresults.
ISO,Geneva(1994).
SeealsoISO5725-5:1998foralternativemethodsofestimatingprecision.
H.
11.
I.
J.
Good,"DegreeofBelief",inEncyclopaediaofStatisticalSciences,Vol.
2,Wiley,NewYork(1982).
H.
12.
J.
Kragten,Analyst,119,2161-2166(1994).
H.
13.
BritishStandardBS6748:1986.
Limitsofmetalreleasefromceramicware,glassware,glassceramicwareandvitreousenamelware.
H.
14.
S.
L.
R.
Ellison,V.
J.
Barwick.
Accred.
Qual.
Assur.
3101-105(1998).
H.
15.
ISO9004-4:1993,TotalQualityManagement.
Part2.
Guidelinesforqualityimprovement.
ISO,Geneva(1993)H.
16.
H.
Kaiser,Anal.
Chem.
4224A(1970).
H.
17.
L.
A.
Currie,Anal.
Chem.
40583(1968).
H.
18.
IUPAC,LimitofDetection,Spectrochim,Acta33B242(1978).
H.
19.
OIMLRecommendationsIR33.
Conventionalvalueoftheresultofweighinginair.
- 不确定学生被ko去世相关文档
- 科索沃学生被ko去世
- 系统工程学生被ko去世
- 患者学生被ko去世
- 警方学生被ko去世
- 短语学生被ko去世
- 核查学生被ko去世
ShockHosting日本机房VPS测试点评
这个月11号ShockHosting发了个新上日本东京机房的邮件,并且表示其他机房可以申请转移到日本,刚好赵容手里有个美国的也没数据就发工单申请新开了一个,这里做个简单的测试,方便大家参考。ShockHosting成立于2013年,目前提供的VPS主机可以选择11个数据中心,包括美国洛杉矶、芝加哥、达拉斯、杰克逊维尔、新泽西、澳大利亚、新加坡、日本、荷兰和英国等。官方网站:https://shoc...
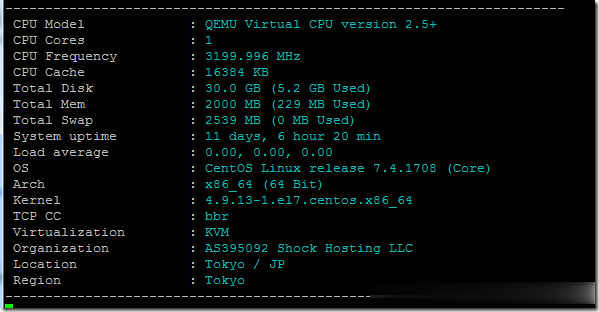
香港云服务器 1核 256M 19.9元/月 Mineserver Ltd
Mineserver(ASN142586|UK CompanyNumber 1351696),已经成立一年半。主营香港日本机房的VPS、物理服务器业务。Telegram群组: @mineserver1 | Discord群组: https://discord.gg/MTB8ww9GEA7折循环优惠:JP30(JPCN2宣布产品可以使用)8折循环优惠:CMI20(仅1024M以上套餐可以使用)9折循...
Sharktech($49/月),10G端口 32GB内存,鲨鱼机房新用户赠送$50
Sharktech 鲨鱼机房商家我们是不是算比较熟悉的,因为有很多的服务商渠道的高防服务器都是拿他们家的机器然后部署高防VPS主机的,不过这几年Sharktech商家有自己直接销售云服务器产品,比如看到有新增公有云主机有促销活动,一般有人可能买回去自己搭建虚拟主机拆分销售的,有的也是自用的。有看到不少网友在分享到鲨鱼机房商家促销活动期间,有赠送开通公有云主机$50,可以购买最低配置的,$49/月的...
学生被ko去世为你推荐
-
flashfxpflashfxp怎么用?dell服务器bios设置dell怎样进入bios设置界面tumblr上不去我家里的网络打不开个别网站社区动力如何大力加强社区基层党组织建设qq挂件QQ免费挂件怎么用图文模块图文模块的标题栏填什么啊?discuzx2DISCUZ X2是PHP还是ASP的?织梦去版权dedecms模板上的版权怎么改成自己想要的版权一键备份一键还原怎么备份?论坛勋章个人论坛的勋章从哪里弄